10 scATAC-seq 分析
scATAC-seq 测的是单细胞层面的染色质可及性:哪些基因组区域在这个细胞里是"打开的",通常对应启动子、增强子等顺式调控元件。和 scRNA-seq 的一个直接区别是稀疏得多——每个细胞只有几千到几万个 fragment,落在 peak 上的更少——所以工具链另成一套。
本节用 10x Genomics 的 PBMC scATAC 10k 数据演示两套主流方案:ArchR(全流程、适合大数据)和 Signac(和 Seurat 无缝衔接,适合已经熟 Seurat 的用户)。
scATAC 能回答 scRNA 不能回答的问题
scRNA-seq 看的是"基因被表达了多少",scATAC-seq 看的是"基因有没有可能被表达"。这两层在很多场景下不一致:
- 一个分化早期的细胞可能"染色质打开了但 mRNA 还没起来"
- 一个静默的基因可能"mRNA 没了但染色质仍开放",意味着该基因还能被快速重新激活
scATAC-seq 适合的核心问题:
- 找驱动转录因子:peak 上做 motif 分析,能直接告诉你"哪个 TF 在这群细胞里�活跃"。RNA 只能看 TF 自己的表达,但 TF 的活性和表达不一定挂钩
- 追踪分化中的"决定时刻":染色质重塑通常先于转录变化,scATAC 能看到 RNA 还没动但命运已定的细胞
- 疾病变异功能化:GWAS 找到的非编码区 SNP,落在哪些细胞类型的开放区域里?这是把"统计关联"翻译成"机制候选"的标准方式
不适合 scATAC:单纯做细胞分型(用 RNA 更稳)、找差异表达(不是它的强项)、稀有亚群定义(数据稀疏,几十个细胞可能不够建 peak)。
分析流水线的关键差异
| 步骤 | scRNA-seq | scATAC-seq |
|---|---|---|
| 原始数据 | counts matrix | fragment file(.tsv.gz) |
| 特征 | 基因 | peak 或 tile |
| 主降维 | PCA | TF-IDF + SVD(称 LSI) |
| 聚类 | 基于 PCA 的 KNN 图 | 基于 LSI 的 KNN 图 |
| 标志特征 | 差异基因 | 差异 peak / motif |
大多数思路是通的:归一化 → 降维 → 找邻居 → 聚类 → 注释。差别在"归一化/降维怎么做"(TF-IDF + SVD)和"特征是什么"(peak 而非基因)。
用 ArchR 做全流程
ArchR 用 Arrow 文件作为底层存储,几十万细胞也能在普通服务器上跑动。
# 初次安装
if (!requireNamespace("devtools", quietly = TRUE)) install.packages("devtools")
devtools::install_github(
"GreenleafLab/ArchR",
ref = "master",
repos = BiocManager::repositories()
)
library(ArchR)
addArchRThreads(threads = 8)
addArchRGenome("hg38") # PBMC 10k 是人类数据
# 从 fragment 文件创建 Arrow(按 sample 分开)
ArrowFiles <- createArrowFiles(
inputFiles = "~/biof3-data/pbmc10k-scatac/atac_fragments.tsv.gz",
sampleNames = "pbmc10k",
minTSS = 4, # TSS enrichment 最低值(典型 > 6 就是好)
minFrags = 1000, # 细胞最低 fragment 数
addTileMat = TRUE,
addGeneScoreMat = TRUE
)
proj <- ArchRProject(ArrowFiles, outputDirectory = "pbmc10k_ArchR")
# 去除 doublet
proj <- addDoubletScores(proj)
proj <- filterDoublets(proj)
minTSS 和 minFrags 是 scATAC 的两条关键 QC 门槛:TSS enrichment 反映信号是否集中在转录起始位点附近,fragment 数衡量测序深度。
降维和聚类走 LSI:
proj <- addIterativeLSI(proj, useMatrix = "TileMatrix", name = "IterativeLSI")
proj <- addUMAP(proj, reducedDims = "IterativeLSI")
proj <- addClusters(proj, reducedDims = "IterativeLSI")
plotEmbedding(proj, colorBy = "cellColData", name = "Clusters")
IterativeLSI 是 ArchR 特色:它会先用 top 可变 tile 做一次 LSI,聚类后再选 top 可变 tile 做第二次——对于稀疏数据这样更稳。
peak 层面的分析
仅靠 tile 做完聚类,真正做差异分析需要先 call peak:
# 为每个 cluster 生成 pseudobulk bigWig,再调用 MACS2
proj <- addGroupCoverages(proj, groupBy = "Clusters")
proj <- addReproduciblePeakSet(proj, groupBy = "Clusters")
proj <- addPeakMatrix(proj)
# 按 cluster 找特征 peak
markerPeaks <- getMarkerFeatures(
ArchRProj = proj,
useMatrix = "PeakMatrix",
groupBy = "Clusters"
)
plotMarkerHeatmap(markerPeaks, cutOff = "FDR <= 0.01 & Log2FC >= 1")
peak 本身只是"开放区域",要看它背后哪些转录因子在起作用,就做 motif 富集:
proj <- addMotifAnnotations(proj, motifSet = "cisbp", name = "Motif")
enrichMotifs <- peakAnnoEnrichment(
seMarker = markerPeaks,
ArchRProj = proj,
peakAnnotation = "Motif",
cutOff = "FDR <= 0.1 & Log2FC >= 0.5"
)
plotEnrichHeatmap(enrichMotifs, n = 7, transpose = TRUE)
每个 cluster 富集出来的 motif 列表是判断"这个 cluster 是什么细胞类型"的重要证据,尤其和 RNA 层面的 marker 基因交叉验证时。
把 ATAC 和 RNA 对齐
如果同时有 scRNA-seq 的参考数据(或来自同一批样本的 Multiome GEX),可以把 RNA 的 cell type 标签"传"到 ATAC 上:
library(Seurat)
rna <- readRDS("pbmc_rna_annotated.rds")
proj <- addGeneIntegrationMatrix(
ArchRProj = proj,
useMatrix = "GeneScoreMatrix",
matrixName = "GeneIntegrationMatrix",
reducedDims = "IterativeLSI",
seRNA = rna,
addToArrow = TRUE,
groupRNA = "cell_type",
nameCell = "predictedCell",
nameGroup = "predictedGroup",
nameScore = "predictedScore"
)
plotEmbedding(proj, colorBy = "cellColData", name = "predictedGroup")
GeneScoreMatrix 是 ArchR 在 peak 基础上估计出的"基因活跃度"代理,跟 RNA 匹配效果通常不错。如果数据是 10x Multiome,RNA 和 ATAC 来自同一细胞,就不用做整合,直接把 barcode 对齐即可。
用 Signac 的版本
Signac 把 ATAC 建模成 Seurat 的 Assay,如果流程已经建在 Seurat �上,衔接会很自然:
library(Signac)
library(Seurat)
counts <- Read10X_h5("~/biof3-data/pbmc10k-scatac/atac_filtered_peak_bc_matrix.h5")
metadata <- read.csv("~/biof3-data/pbmc10k-scatac/atac_singlecell.csv", row.names = 1)
chrom_assay <- CreateChromatinAssay(
counts = counts,
sep = c(":", "-"),
genome = "hg38",
fragments = "~/biof3-data/pbmc10k-scatac/atac_fragments.tsv.gz",
min.cells = 10,
min.features = 200
)
pbmc <- CreateSeuratObject(chrom_assay, assay = "peaks", meta.data = metadata)
pbmc <- NucleosomeSignal(pbmc)
pbmc <- TSSEnrichment(pbmc, fast = FALSE)
pbmc <- RunTFIDF(pbmc)
pbmc <- FindTopFeatures(pbmc, min.cutoff = "q0")
pbmc <- RunSVD(pbmc)
pbmc <- RunUMAP(pbmc, reduction = "lsi", dims = 2:30)
pbmc <- FindNeighbors(pbmc, reduction = "lsi", dims = 2:30)
pbmc <- FindClusters(pbmc, algorithm = 3, verbose = FALSE)
DimPlot(pbmc, label = TRUE) + NoLegend()
dims = 2:30 是 Signac 教程里的一个 convention:LSI 的第一个维度通常和测序深度高度相关,跳过它能避免聚类被测序深度主导。
真实示例:PBMC 10k scATAC 走 Signac
配套脚本 module11_scatac_sci.R 用 10x Genomics 的 PBMC 10k scATAC v2 数据(filtered peak matrix + singlecell.csv,约 200 MB)走一遍 Signac 的标准流程。首次运行会从 10x 官网下载数据到 ~/biof3-data/pbmc10k-scatac/。
Rscript scripts/single-cell/sc11_scatac_sci.R
脚�本顺序:Read10X_h5 读 peak matrix → CreateChromatinAssay 建对象 → 基于 singlecell.csv 里的 QC 指标过滤 → RunTFIDF + RunSVD(LSI)→ UMAP + Leiden 聚类 → 差异 peak → 可视化。
每张图看什么
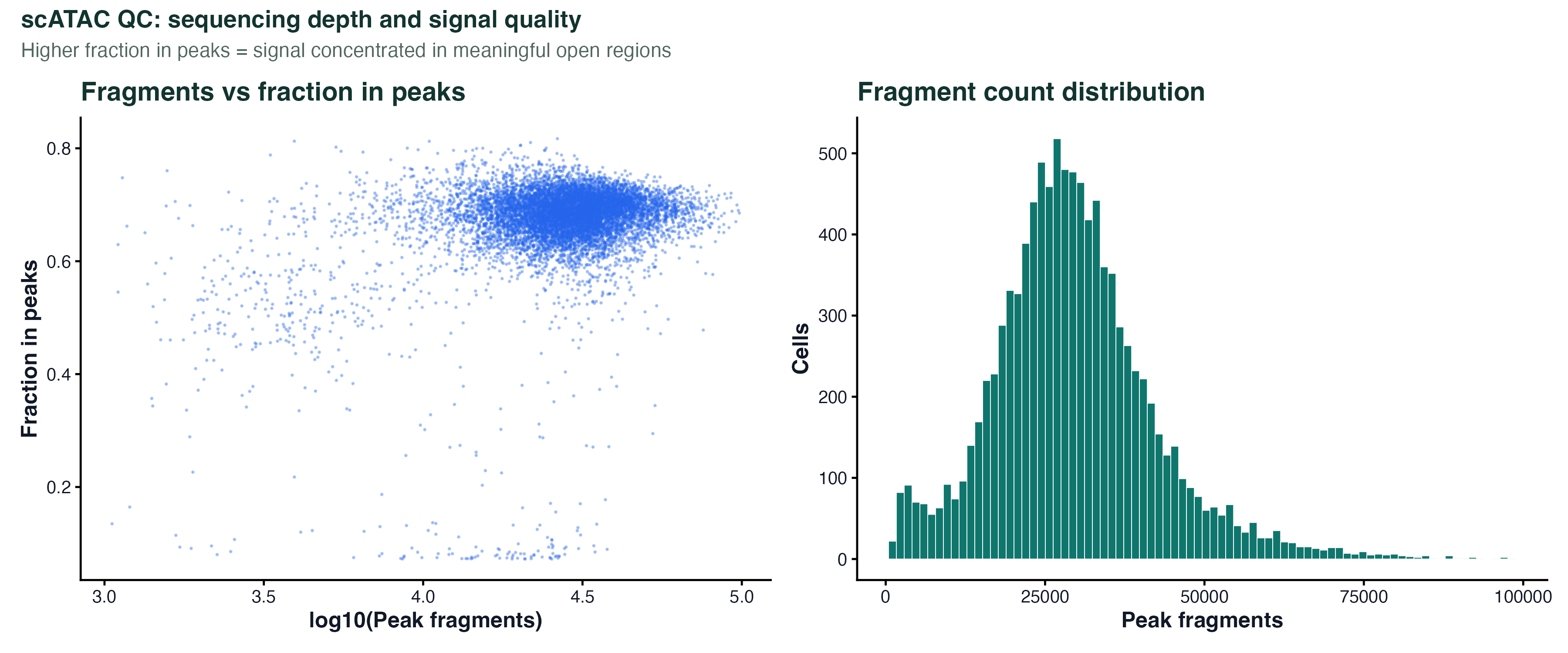
图 1:左图是每个细胞的 peak fragment 数 vs 落在 peak 区域的 fragment 占比。占比越高说明信号越集中在有意义的开放区域(而不是随机噪声)。右图是 fragment 数的直方图,确认大部分细胞在合理范围内。
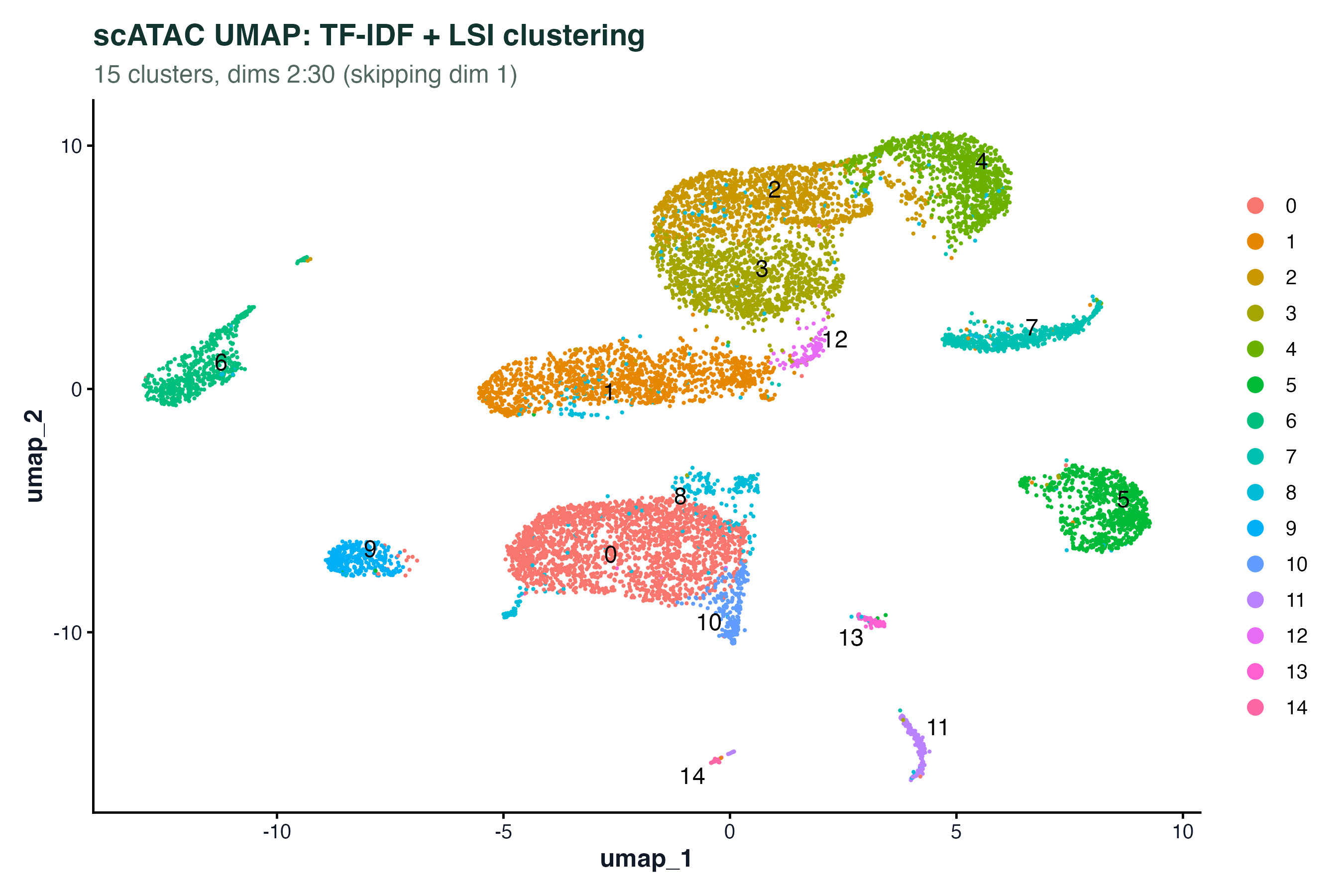
图 2:TF-IDF + LSI 降维后在 UMAP 上的聚类。15 个 cluster 对应 PBMC 里的主要免疫细胞类型。和 scRNA-seq 的 UMAP 相比,scATAC 的 cluster 边界通常更模糊(因为信号更稀疏),但大类分离仍然清晰。
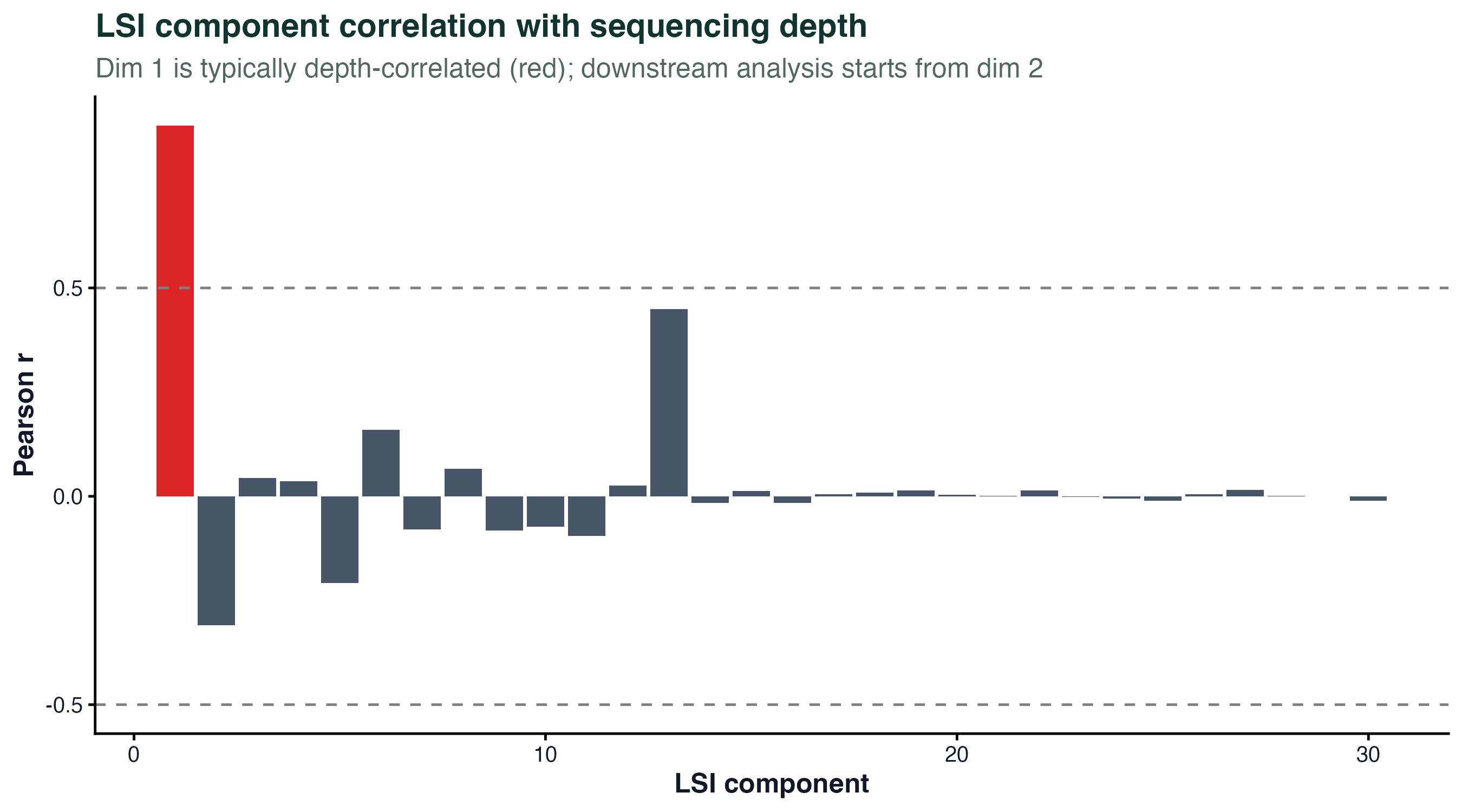
图 3:LSI 各维度与测序深度的 Pearson 相关。第 1 维和深度高度相关(红色柱子),所以下游分析从 dim 2 开始。如果第 2 维也和深度相关,说明 TF-IDF 归一化没完全消除深度效应,需要检查 QC 是否够严。
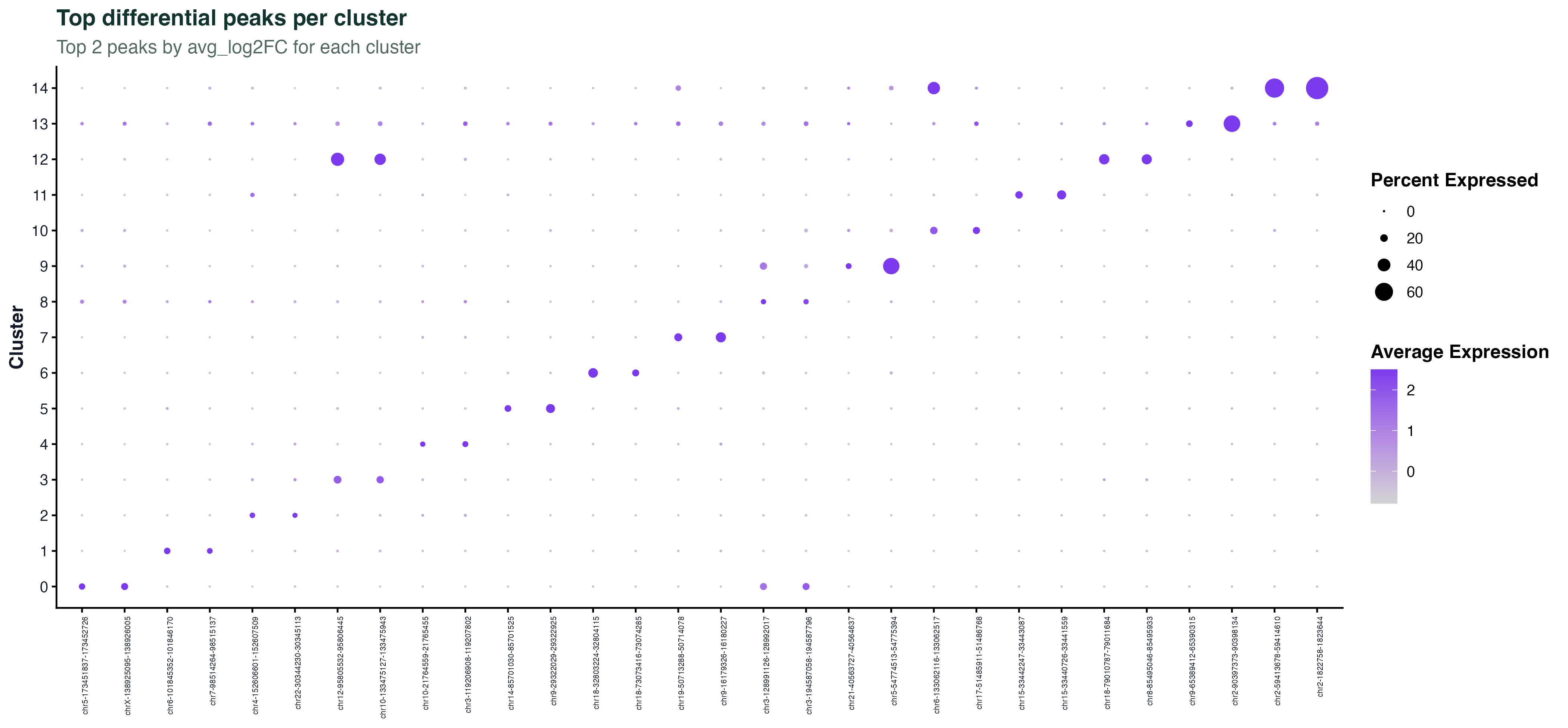
图 4:每个 cluster 取 avg_log2FC 最高的 2 个差异 peak,画 dotplot。颜色代表平均可及性,点大小代表阳性细胞比例。和 scRNA-seq 的差异基因 dotplot 读法一样,只是横轴变成了基因组坐标(chr:start-end)。
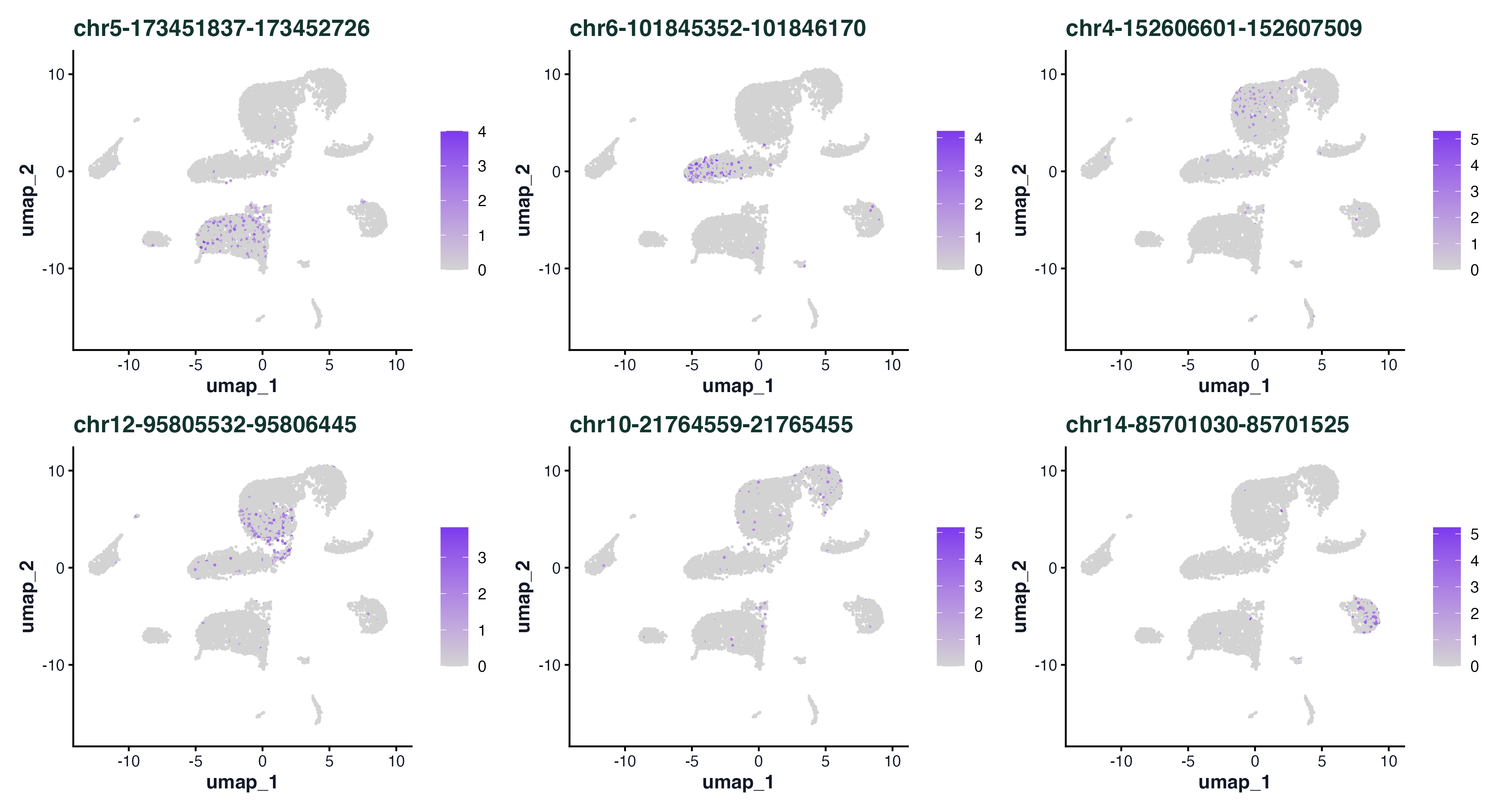
图 5:前 6 个 cluster 各自最强的差异 peak 在 UMAP 上的可及性分布。每张子图里亮色区域就是"这个 peak 在哪些细胞里打开了"。如果一个 peak 落在某个已知基因的启动子上,就能直接推测这个 cluster 的身份。
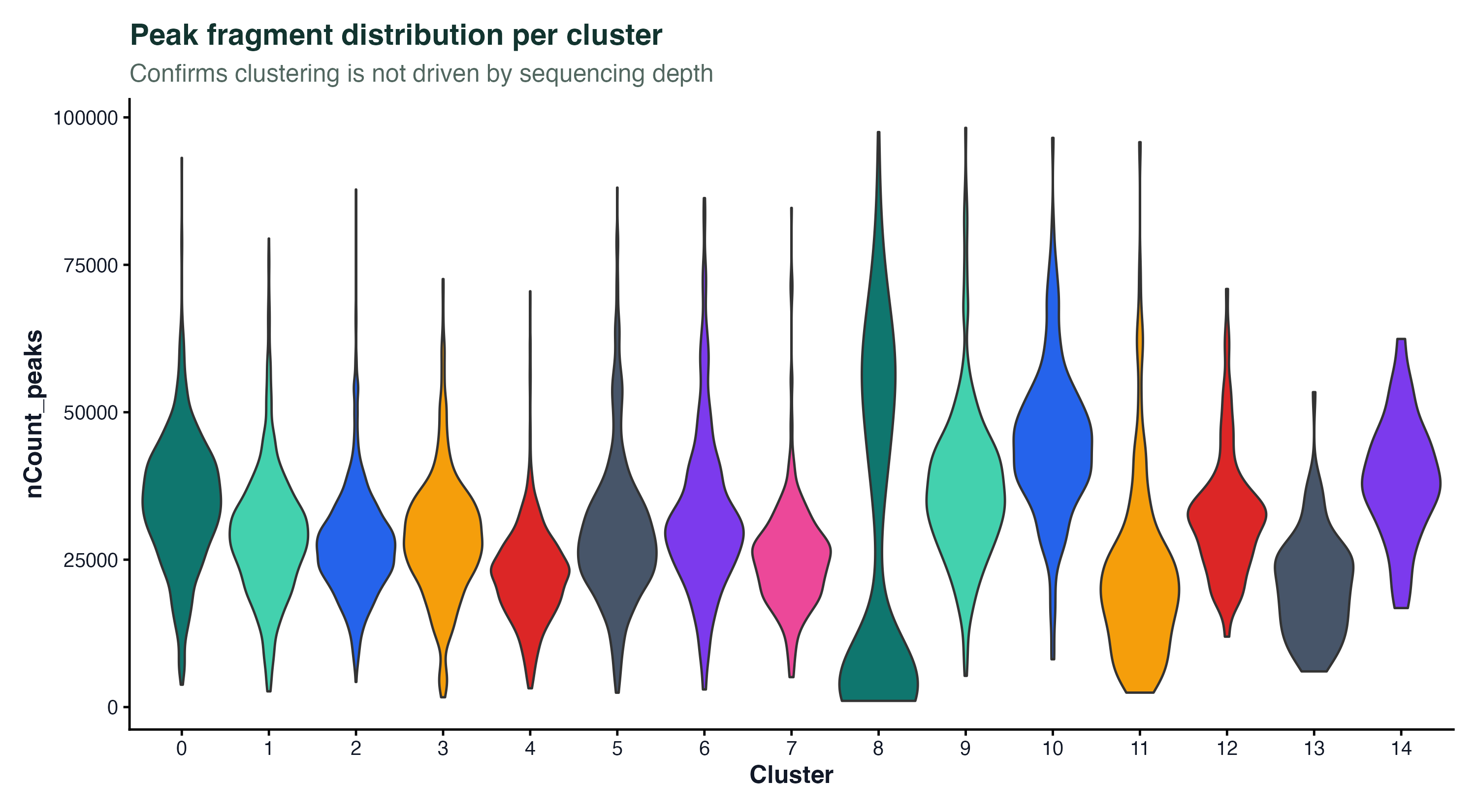
图 6:各 cluster 的 peak fragment 数分布。确认聚类不是被测序深度驱动的——如果某个 cluster 的深度明显偏高或偏低,说明聚类可能有技术偏差。这张图里各 cluster 深度分布基本一致,说明聚类反映的是真实的生物学差异。
套到自己数据上
脚本只需要 filtered_peak_bc_matrix.h5 和 singlecell.csv,不需要 fragment 文件(省掉 2.6 GB 下载)。如果你有 fragment 文件,可以额外加 NucleosomeSignal + TSSEnrichment 做更精细的 QC,以及用 GeneActivity 估计基因活跃度来辅助注释。把脚本里的 URL 换成自己��的数据路径即可。
工具选择建议
- 只做单样本、想用现成 Seurat 流程 → Signac
- 多样本、要出差异 peak + motif 报告 → ArchR
- 10x Multiome(同时有 RNA + ATAC)→ Seurat v5 自带
IntegrateLayers,或者用MultiVI
常见坑
坑 1:LSI 第 1 维和测序深度高度相关
scATAC 数据里,LSI 的 dim1 通常代表"细胞测了多少 fragment"而不是生物学差异。下游分析从 dim2 开始(dims = 2:30),不然聚类会被深度主导。这是 scATAC 和 scRNA 在用 PCA / LSI 上最重要的一个差别。
坑 2:用 cluster 内细胞 < 100 去 call peak
addReproduciblePeakSet 默认要求每个 group 至少 ~25 个细胞做 pseudobulk。< 100 时 peak 会非常嘈杂,一些真实 peak 测不到、随机噪声却被叫成 peak。小 cluster 要么合并相近的,要么先用全细胞 call 一套 peak 再分组分析。
坑 3:Gene Activity Score 当成基因表达量解读
GeneActivity 把 peak 信号映射到基因(按距离加权),是"这个基因附近染色质开放程度"的代理,不是 RNA 表达量。两者在大部分情况下相关但不一致。做差异分析、富集分析时 GeneActivity 当代理用可以,但写论文时要说清楚是 ATAC 推断的活性、不是 RNA 表达。
坑 4:跨样本整合时忘了用同一套 peak
每个样本各自 call peak 得到的 peak set 不一样,多样本整合需要先 merge 成统一 peak set。用各样本独立 peak set 直接合并矩阵是错的 — 同一基因组位置在不同样本里被切成不同 peak,矩阵里看上去就是"该样本特有"。ArchR 的 addReproduciblePeakSet + addPeakMatrix 自动处理;Signac 用 reduce() + FeatureMatrix 重算。
坑 5:fragment 文件没建索引就跑
Signac 的很多函数(NucleosomeSignal、TSSEnrichment、CoveragePlot)需要 fragments.tsv.gz.tbi 索引文件。只下了 .tsv.gz 没下 .tbi 时调用会报"file not found"。要么一起下,要么自己 tabix -p bed fragments.tsv.gz 建一个。
下载资源
下一步
接着深入(已是单细胞专题最后一篇):
- bulk RNA-seq 实践教程 — 单细胞已经走完,回到 bulk 学习"群体基因表达"的另一面
- 表观组学实践教程 — bulk ATAC、ChIP-seq、甲基化等更深入的染色质分析
横向延伸:
- 07 多模态数据分析 — 10x Multiome 把 RNA + ATAC 测在同一细胞,整合分析比独立 scATAC 信息量大很多
- 03 质量控制、聚类与细胞类型注释 — scATAC 的 cell type 注释通常借助配套 scRNA 参考,回头复习 scRNA 注释流程
参考资源
离线资料下载
手册 HTML / PDF 已在后台预生成,点击后直接下载网站静态资源。