06 细胞-细胞通讯分析
单细胞数据让我们能拿到组织里每种细胞各自在表达什么,但很多生物学问题的答案不在某一类细胞里,而在"A 类细胞的表达信号传到 B 类细胞上引发了什么"。细胞-细胞通讯分析(cell-cell communication,或 ligand-receptor analysis)就是从这个角度建模:根据每类细胞里配体(ligand)和受体(receptor)的表达水平,结合已知的 L-R 相互作用数据库,推测哪些细胞对之间在"说话"。
本章用三种主流工具走一遍:CellChat(R,目前最主流)、CellPhoneDB(Python,数据库最严谨)、NicheNet(R,往下游推信号效应)。它们背后的思路一致,区别在配套数据库、建模假设和输出形式。
这类分析能回答什么
- 肿瘤细胞上哪些配体最可能在驱动 T 细胞耗竭
- 感染后巨噬细胞向上皮细胞发出哪种炎症信号
- 发育过程中一��个 niche 内的 stem cell 在接收哪些信号
- 治疗前后细胞通讯网络整体改变在哪
注意它给的是"可能有通讯"的证据,不是直接验证过的通讯。配体受体共表达只是必要条件,不是充分条件;把某个候选结论当作定论之前,需要实验或文献二次验证。
这套分析的核心假设要先看清楚
所有 L-R 工具都在做同一件事:配体在 A 类细胞高表达 + 受体在 B 类细胞高表达 → 推测 A 给 B 发信号。这个推断里有几个常被忽视的前提:
| 假设 | 现实情况 |
|---|---|
| mRNA 表达 = 蛋白存在 | 配体 mRNA 高,蛋白可能未分泌(翻译后调控) |
| 共表达 = 物理接触 | 同一组织里也有跨距离的扩散,更别说不同组织 |
| 配体只激活已知受体 | 一个配体常能激活多个受体,反之亦然 |
| 数据库里的 L-R 对都对 | 数据库整理参差,有些相互作用证据不强 |
结论不能止步于工具输出:看到"WNT5A 从巨噬到 T 细胞"的预测后,还要看 WNT5A 蛋白能不能分泌、T 细胞 FZD5 是不是真的在该 cluster 里高表达(不只是 marker 检测到)、文献里这条通路在该场景下是否报道过。这是把"工具结果"翻译成"生物学结论"必经的步骤。
工具选择
| 工具 | 语言 | 数据库 | 特点 |
|---|---|---|---|
| CellChat | R | 自建、分类清晰 | 可视化最完整、比较分析成熟 |
| CellPhoneDB | Python | 人工整理,复合物级 | 对异源二聚体受体处理好 |
| NicheNet | R | 多源整合 + 下游网络 | 从配体推到目标基因表达变化 |
| LIANA | R / Py | 集成多种方法 | 不确定哪个方法更好时拿来对比 |
新项目从 CellChat 开始。成熟、教程全、可视化够用。
CellChat:完整流程
安装和读入
# CellChat 的维护仓库已经从 sqjin 迁到 jinworks
devtools::install_github("jinworks/CellChat")
library(CellChat)
library(patchwork)
library(Seurat)
cellchat <- createCellChat(object = seurat_obj, group.by = "cell_type")
如果对象不是 Seurat,也可以手动构造:
data.input <- GetAssayData(seurat_obj, assay = "RNA", slot = "data")
meta <- seurat_obj@meta.data
cellchat <- createCellChat(object = data.input, meta = meta, group.by = "cell_type")
加载 L-R 数据库
# 人
CellChatDB <- CellChatDB.human
# 鼠用 CellChatDB.mouse
# 查看分类(Secreted Signaling / Cell-Cell Contact / ECM-Receptor)
showDatabaseCategory(CellChatDB)
cellchat@DB <- CellChatDB
# 也可以只用某一子集
# cellchat@DB <- subsetDB(CellChatDB, search = "Secreted Signaling")
预处理和通讯概率计算
cellchat <- subsetData(cellchat)
cellchat <- identifyOverExpressedGenes(cellchat)
cellchat <- identifyOverExpressedInteractions(cellchat)
cellchat <- computeCommunProb(cellchat, type = "triMean", trim = 0.1)
cellchat <- filterCommunication(cellchat, min.cells = 10)
cellchat <- computeCommunProbPathway(cellchat)
cellchat <- aggregateNet(cellchat)
computeCommunProb 是核心:它把配体和受体在各类细胞里的表达量代入一个 mass-action 模型,估计每对 "源细胞-目标细胞" 在每个 L-R 对上的通讯概率。filterCommunication 过掉细胞数太�少的群体(默认 10)。
全局网络可视化
groupSize <- as.numeric(table(cellchat@idents))
par(mfrow = c(1, 2), xpd = TRUE)
netVisual_circle(
cellchat@net$count,
vertex.weight = groupSize, weight.scale = TRUE, label.edge = FALSE,
title.name = "Number of interactions"
)
netVisual_circle(
cellchat@net$weight,
vertex.weight = groupSize, weight.scale = TRUE, label.edge = FALSE,
title.name = "Interaction strength"
)
两张图一起看:数量图显示"有多少条通讯路径",强度图显示"这些路径加起来信号有多强"。新项目里通常先看强度图。
单个细胞类型的通讯
看某一种细胞(例如巨噬细胞)分别跟其他细胞类型之间的通讯强度:
mat <- cellchat@net$weight
par(mfrow = c(2, 2), xpd = TRUE)
for (i in seq_len(nrow(mat))) {
mat2 <- matrix(0, nrow = nrow(mat), ncol = ncol(mat), dimnames = dimnames(mat))
mat2[i, ] <- mat[i, ]
netVisual_circle(
mat2,
vertex.weight = groupSize, weight.scale = TRUE,
edge.weight.max = max(mat), title.name = rownames(mat)[i]
)
}
按信号通路可视化
CellChat 会把 L-R 对聚�合到已知信号通路(WNT、TGFβ、TNF 等)层面:
cellchat@netP$pathways # 看被激活的通路列表
pathways.show <- c("WNT")
netVisual_aggregate(cellchat, signaling = pathways.show, layout = "hierarchy")
netVisual_aggregate(cellchat, signaling = pathways.show, layout = "circle")
netVisual_aggregate(cellchat, signaling = pathways.show, layout = "chord")
netVisual_heatmap(cellchat, signaling = pathways.show, color.heatmap = "Reds")
单个 L-R 对
想具体看"WNT 通路里哪一对 L-R 贡献最大":
netAnalysis_contribution(cellchat, signaling = pathways.show)
pairLR.WNT <- extractEnrichedLR(
cellchat, signaling = pathways.show, geneLR.return = FALSE
)
netVisual_individual(
cellchat, signaling = pathways.show,
pairLR.use = "WNT5A_FZD5", layout = "hierarchy"
)
识别 sender / receiver 角色
每类细胞在通讯网络里是主要发送者还是主要接收者?
cellchat <- netAnalysis_computeCentrality(cellchat, slot.name = "netP")
netAnalysis_signalingRole_network(
cellchat, signaling = pathways.show,
width = 8, height = 2.5, font.size = 10
)
ht1 <- netAnalysis_signalingRole_heatmap(cellchat, pattern = "outgoing", width = 8, height = 10)
ht2 <- netAnalysis_signalingRole_heatmap(cellchat, pattern = "incoming", width = 8, height = 10)
ht1 + ht2
识别整体通讯模式
把所有通路聚成若干模式,看有没有"一组细胞集体往外发某一组信号":
cellchat <- identifyCommunicationPatterns(cellchat, pattern = "outgoing", k = 3)
cellchat <- identifyCommunicationPatterns(cellchat, pattern = "incoming", k = 3)
netAnalysis_river(cellchat, pattern = "outgoing")
netAnalysis_river(cellchat, pattern = "incoming")
真实示例:PBMC 3k 上跑一次 CellChat
继续用模块 05 的那份 PBMC 3k 数据(同样的 QC、marker-based 粗注释),这次不做轨迹,而是跑一遍 CellChat。下面的六张图来自配套脚本 module07_cellchat_sci.R 在真实数据上的输出:
Rscript scripts/single-cell/sc07_cellchat_sci.R
脚本做的事和上面讲的流程完全一致:创建对象、加载 CellChatDB.human、算过表达 L-R 对、推断通讯概率、过滤低细胞数的群体、在通路层面汇聚、计算 sender/receiver centrality,最后按这六个角度出图。PBMC 3k 是健康外周血,细胞间通讯不如肿瘤微环境剧烈,但 T/B/NK/monocyte/DC 之间的 MHC-II、APP、CD45、CXCR/CCR 一族信号足够当教学演示。
每张图看什么
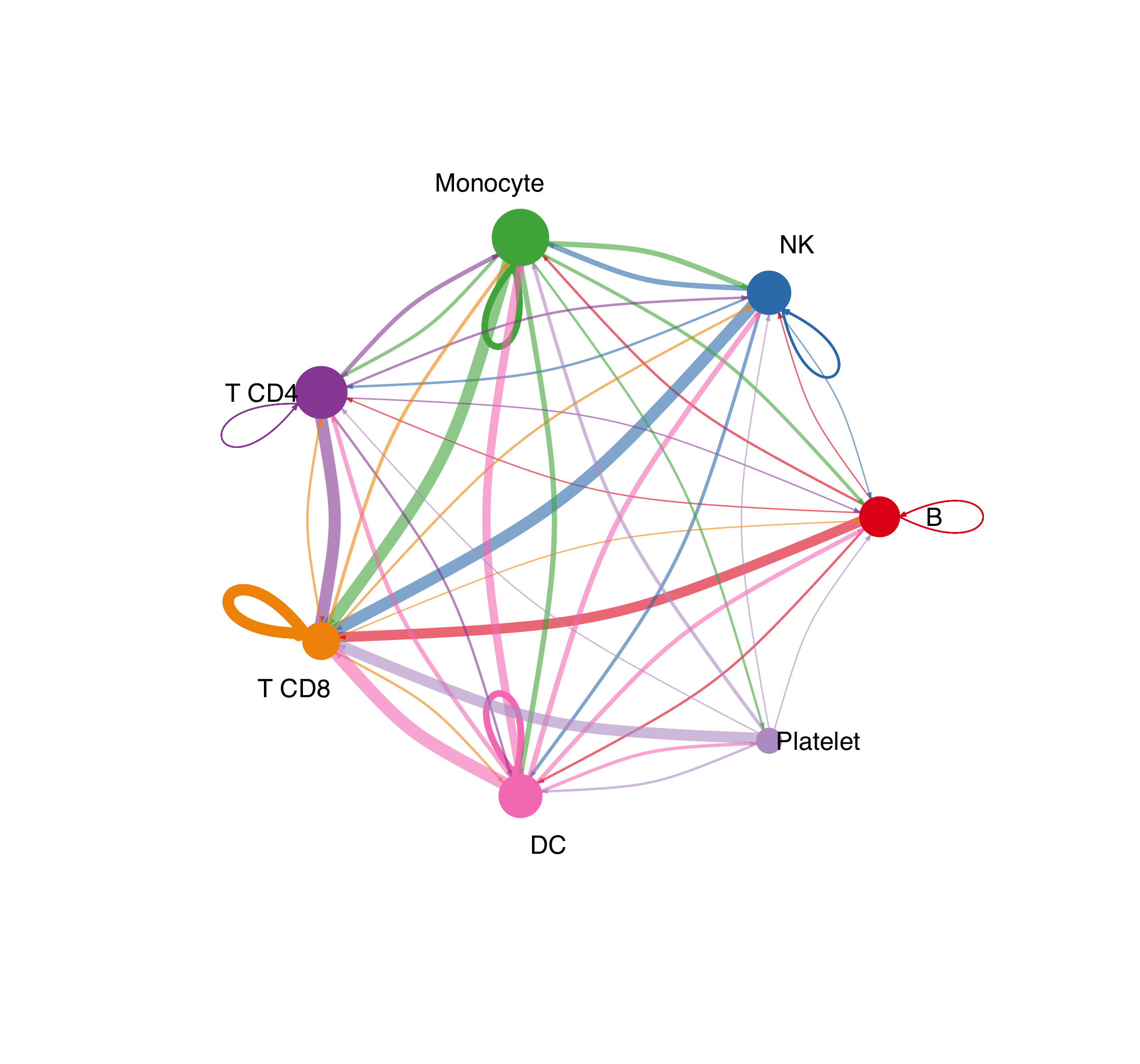
图 1:每对细胞类型之间有多少条显著的 L-R 通讯路径(节点大小反映该类细胞的数量)。这张图回答"谁跟谁有话说",但不告诉你信号有多强。
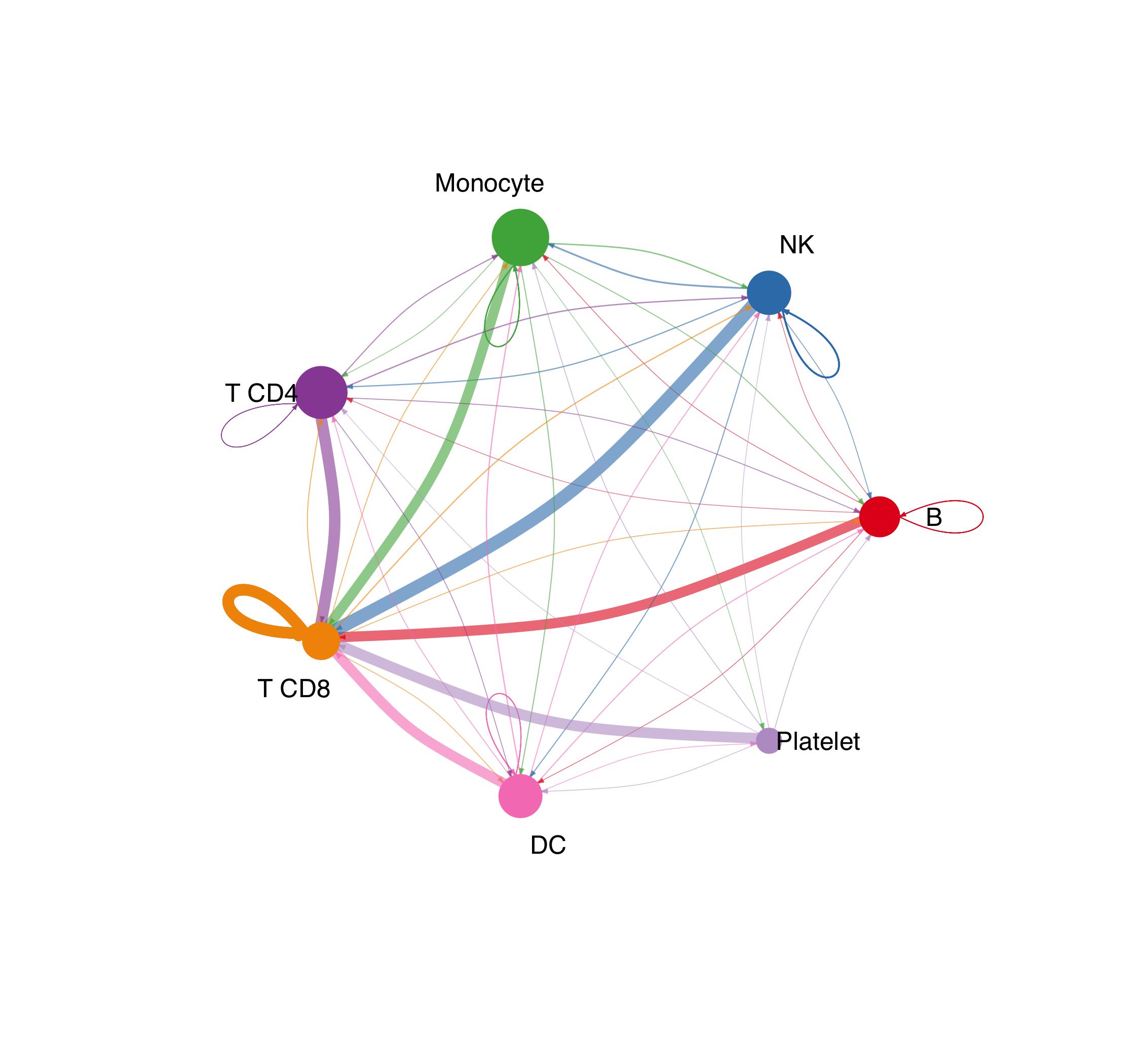
图 2:把每对之间的通讯概率之和画出来。数量 vs 强度经常给出不同的直觉:两个细胞类型之间可能条数不多,但每条都很强。
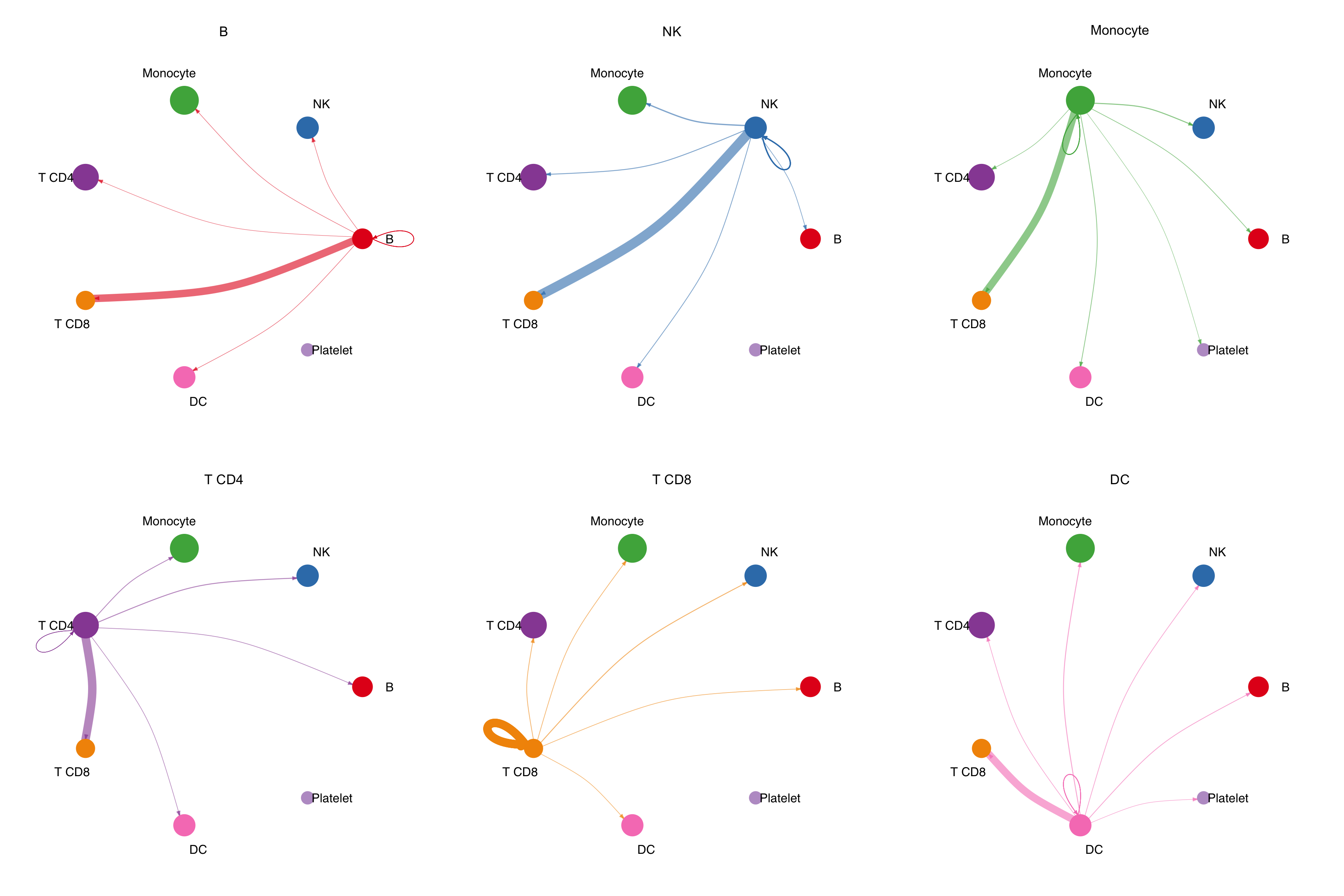
图 3:把"通讯强度圆圈图"按发送方拆成六张小图。每张小图显示某类细胞向其他细胞发出的信号。真实项目里常用来回答"单核细胞具体在向谁说话"。
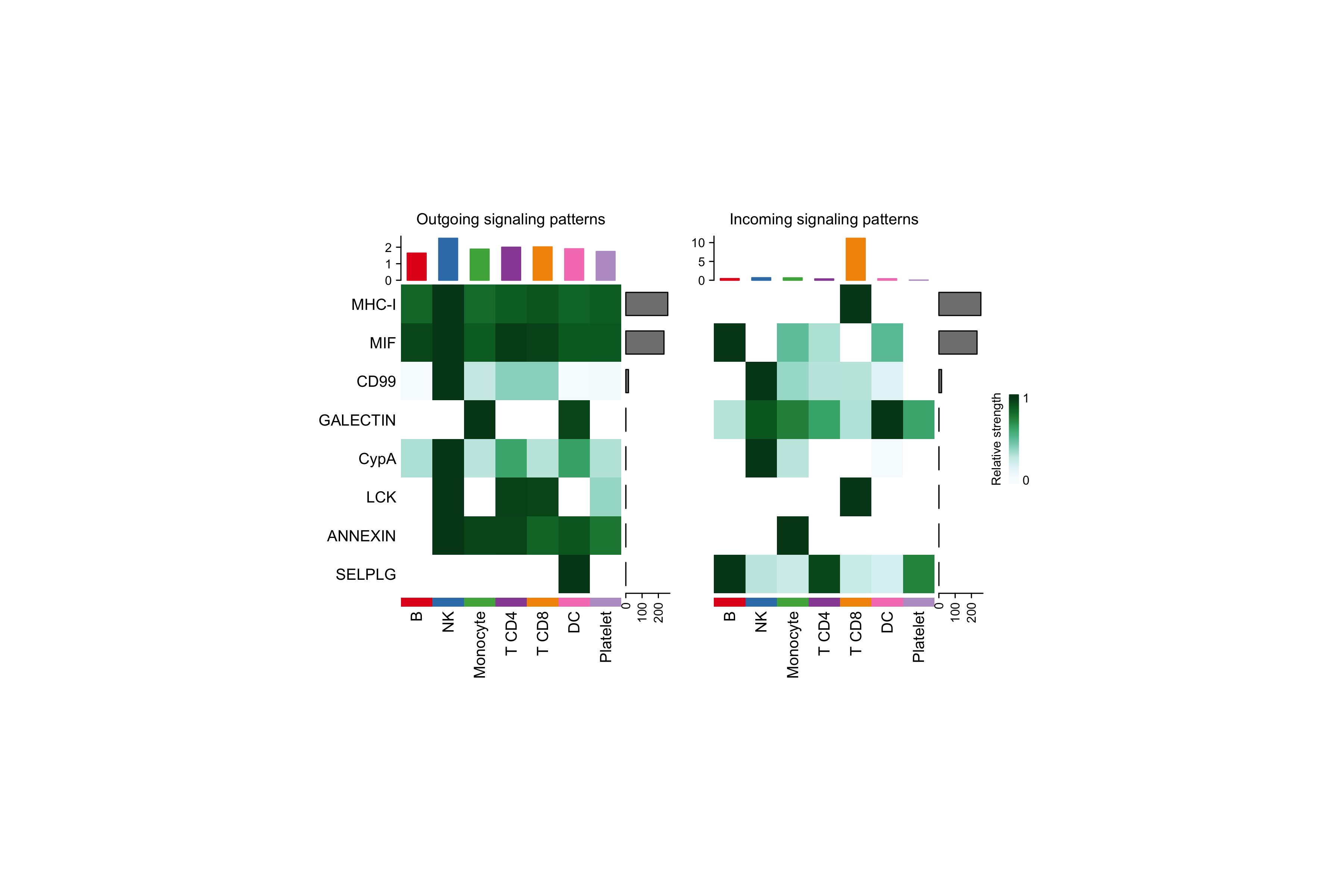
图 4:左图每行是一条通路、每列是一类细胞;颜色深浅代表该类细胞作为发送方在该通路上的贡献。右图是同样的矩阵但代表接收方。从这张图可以快速看出"哪类细胞发出哪些信号"、"哪类细胞最能接到哪些信号"。
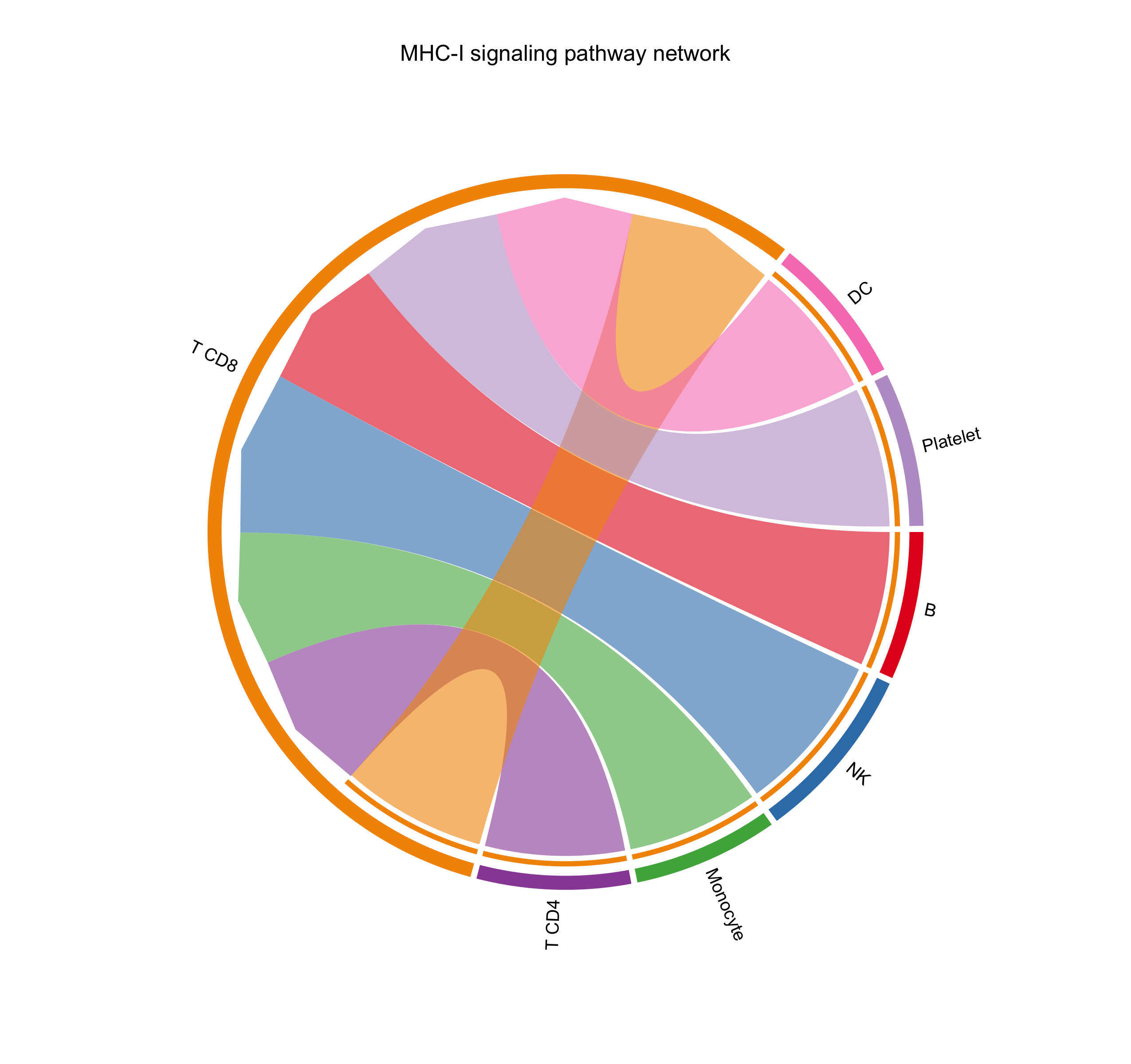
图 5:选一条在 PBMC 里最稳健的通路(优先 MHC-II;没有就退回到 CellChat 检测到��的第一条),用 chord 图显示这条通路上每对细胞的通讯。chord 图的好处是"细胞对"和"信号强度"同屏可读。
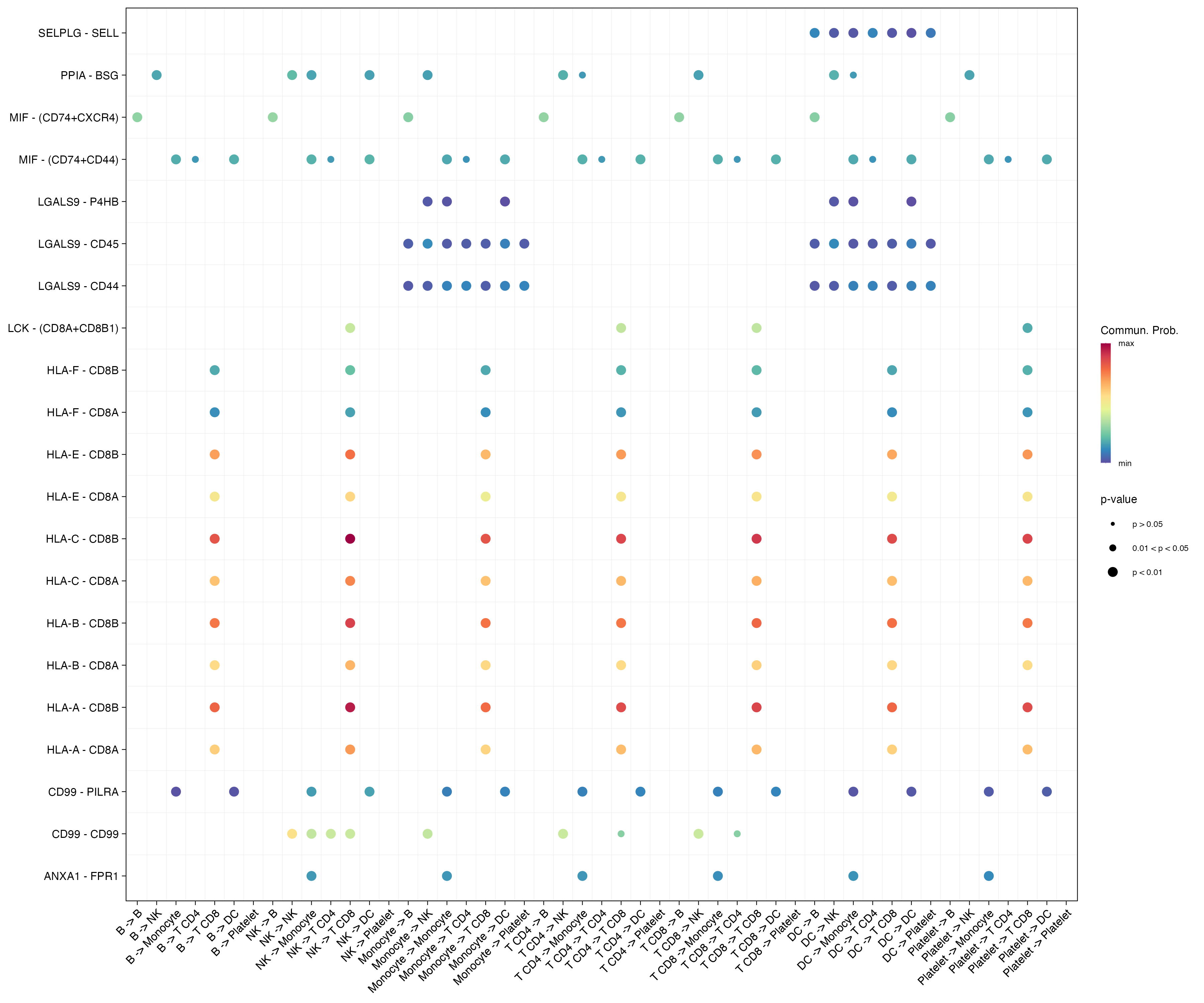
图 6:每个气泡是一个"源细胞 → 目标细胞"上某个 L-R 对的通讯,颜色代表通讯概率、大小代表显著性 p 值。把前六张图看完后,具体哪些 L-R 对在驱动结论,要回到这张图确认。
把流程套到自己的数据上
把脚本第 80-120 行(读 Seurat 对象 + 做 marker 粗注释)换成自己已经注释好的 Seurat 对象和对应的 cell type 列就行。其余的 CellChat 调用几乎不用改。真实项目里特别要关注:
- 细胞数少的 cluster(< 10)会被
filterCommunication丢掉,改min.cells之前先看table(cellchat@idents) computeCommunProb默认用triMean,稀疏数据里过于保守。样本数少 cluster 小的项目可以试type = "truncatedMean", trim = 0.1让结果更丰富- 想比较两个条件(治疗 vs 对照),参考下面"对照条件下的比较分析"小节,用
mergeCellChat+compareInteractions
CellPhoneDB:Python 替代
CellPhoneDB 的数据库对复合物受体(比如 IL-12 受体是 IL12RB1+IL12RB2 的二聚体)处理得最规范。样本之间做跨病人比较时它的置换检验更稳妥。
conda create -n cellphonedb python=3.8
conda activate cellphonedb
pip install cellphonedb
先从 Seurat 导出 counts 和 metadata:
write.table(
as.matrix(seurat_obj@assays$RNA@data),
"counts.txt", sep = "\t", quote = FALSE
)
meta_data <- data.frame(
Cell = rownames(seurat_obj@meta.data),
cell_type = seurat_obj$cell_type
)
write.table(meta_data, "meta.txt", sep = "\t", quote = FALSE, row.names = FALSE)
命令行跑:
cellphonedb method statistical_analysis \
meta.txt counts.txt \
--counts-data=gene_name \
--threads=4
输出在 out/pvalues.txt 和 out/means.txt。可视化可以用自带工具,也可以自己画:
import pandas as pd
import seaborn as sns
import matplotlib.pyplot as plt
means = pd.read_csv("out/means.txt", sep="\t")
pvalues = pd.read_csv("out/pvalues.txt", sep="\t")
plt.figure(figsize=(15, 10))
sns.heatmap(means, cmap="viridis", cbar_kws={"label": "Mean expression"})
plt.tight_layout()
NicheNet:从配体推到下游目标基因
前面两种工具回答"谁在跟谁通讯"。NicheNet 更进一步:给定接收细胞里的一组差异基因,推测哪些配体最可能是导致这些基因变化的"上游驱动"。
devtools::install_github("saeyslab/nichenetr")
library(nichenetr)
library(tidyverse)
# NicheNet 的三个核心矩阵,作者已经预计算好
ligand_target_matrix <- readRDS(url("https://zenodo.org/record/3260758/files/ligand_target_matrix.rds"))
lr_network <- readRDS(url("https://zenodo.org/record/3260758/files/lr_network.rds"))
weighted_networks <- readRDS(url("https://zenodo.org/record/3260758/files/weighted_networks.rds"))
定义 sender 和 receiver,提取各自的表达基因:
sender_celltypes <- c("CD4 T", "CD8 T")
receiver <- "B"
expressed_genes_receiver <- get_expressed_genes(receiver, seurat_obj, pct = 0.10)
expressed_genes_sender <- get_expressed_genes(sender_celltypes, seurat_obj, pct = 0.10)
background_expressed_genes <- union(expressed_genes_receiver, expressed_genes_sender)
接收细胞里感兴趣的基因(例如差异基因)作为 geneset_oi:
geneset_oi <- c("CD69", "CD44", "IL2RA") # 换成你自己的差异基因列表
potential_ligands <- intersect(unique(lr_network$from), expressed_genes_sender)
ligand_activities <- predict_ligand_activities(
geneset = geneset_oi,
background_expressed_genes = background_expressed_genes,
ligand_target_matrix = ligand_target_matrix,
potential_ligands = potential_ligands
)
best_upstream_ligands <- ligand_activities |>
top_n(20, pearson) |>
arrange(-pearson) |>
pull(test_ligand)
这给你的是"最可能解释 receiver 里那组基因变化的 20 个候选配体"。配合实验验证是很合理的第一步筛选。
对照条件下的比较分析
治疗 vs 对照、健康 vs 疾病这种比较,CellChat 有现成流程:
# 假设你已分别做好两个 CellChat 对象
cellchat_list <- list(Control = cellchat_control, Treated = cellchat_treated)
cellchat_merged <- mergeCellChat(cellchat_list, add.names = names(cellchat_list))
# 总体通讯量对比
gg1 <- compareInteractions(cellchat_merged, show.legend = FALSE, group = c(1, 2))
gg2 <- compareInteractions(cellchat_merged, show.legend = FALSE, group = c(1, 2), measure = "weight")
gg1 + gg2
# 哪些细胞对之间的通讯变化最大
netVisual_diffInteraction(cellchat_merged, weight.scale = TRUE)
# 哪些通路在两个条件间差异最大
rankNet(cellchat_merged, mode = "comparison", stacked = TRUE, do.stat = TRUE)
肿瘤微环境的一个典型问题
T 细胞耗竭和免疫检查点通路是肿瘤免疫里最常看的方向。分析流程上没有特别之处,但有几个要关注的点:
- 可视化时把
sources.use = "Tumor"和targets.use = c("CD4 T", "CD8 T"),专门看肿瘤到 T 的通讯 - 关注 PD-L1/PD-1、CTLA4/CD80、TIGIT/PVR、LAG3 这几组
- CellChat 把这几个都归到 "Immune Checkpoint" 类别下,用
showDatabaseCategory查得到
netVisual_bubble(
cellchat,
sources.use = "Tumor",
targets.use = c("CD4 T", "CD8 T"),
remove.isolate = FALSE
)
netVisual_aggregate(cellchat, signaling = "PD-L1", layout = "hierarchy")
几个容易被忽视的问题
- 配体和受体表达的阈值很影响结果。不同方法默认的最低表达百分比不一样,小 cluster 特别容易被过滤掉。
- 同义词和 isoform。数据库里配体名和你矩阵里的 gene symbol 可能对不上,运行前最好抽几个已知的手工核对一下。
- "通讯概率" 不是"通讯发生"。它是个相对量,跨数据集不能直接比。
- 空间信息被忽略了。方法都假设所有细胞都能接触到所有其他细胞;真实组织里很多通讯是局部的。如果数据是空间转录组,应该结合空间分析做通讯推断。
常见坑
坑 1:cluster 数太少 / 太多都不能跑
CellChat 的统计基础是"每个 group 内的细胞数足够估计平均表达"。< 10 个细胞会被 filterCommunication 自动丢掉,但 10-30 个仍然不可靠(结果对单细胞的离群值敏感)。通讯分析前合并 cluster,每组至少 50 个细胞比较稳。
坑 2:把"通讯概率"当成"通讯量"做跨数据集比较
CellChat 的概率值是相对量,两个数据集之间直接比绝对值是错的。要比较组间差异必须用 mergeCellChat + compareInteractions,让工具按相对差异处理。
坑 3:分泌信号 / 接触信号混在一起看
CellChatDB 把 L-R 对分成 Secreted Signaling、Cell-Cell Contact、ECM-Receptor 三类。这三类的生物学含义完全不同:分泌信号能跨距离,接触信号要细胞物理相邻,ECM 要看基质成分。先用 subsetDB(CellChatDB, search = "Secreted Signaling") 选定一类再分析,结论才有意义。
坑 4:忽略复合物受体,只看单基因受体
很多免疫受体是多亚基复合物(IL-12R = IL12RB1 + IL12RB2、整合素 = α + β),单看其中一个基因高表达不代表受体功能位。CellPhoneDB 对复合物处理最严谨,做免疫学项目优先用它。
坑 5:把单条 L-R 对的结论当真
CellChat 一次跑出几百到几千条 L-R 对,单条 p < 0.05 不稀奇。只信"重复出现在多条 L-R 对的同一通路":比如同一对细胞间 WNT5A-FZD5、WNT5A-ROR2、WNT5A-FZD7 都显著,结论才比单条可靠。
下载资源
下一步
接着深入:
- 07 多模态数据分析 — 蛋白层面的受体表达直接来自 ADT 测量,做通讯分析比 RNA 推断更可靠
- 09 空间转录组学 — 在空间数据上做通讯,能直接看到 sender 和 receiver 的物理距离
横向延伸:
- 05 轨迹推断与拟时序分析 — 把通讯结果和分化轨迹结合,看哪些信号驱动了状态切换
- Armingol 2021 通讯方法综述 — 系统对比各类工具的假设和适用范围
参考资源
离线资料下载
手册 HTML / PDF 已在后台预生成,点击后直接下载网站静态资源。