09 空间转录组学
空间转录组在 scRNA-seq 的基础上保留了一个关键维度:每条表达信号来自切片的哪个位置。不同技术的空间分辨率差别很大:10x Visium 把组织打在微珠芯片上,每个 spot 约 55 μm 包含数个细胞;Slide-seq、MERFISH、seqFISH+ 能做到单细胞或亚细胞分辨率,代价是数据量更大、分析工具更专。
本节用 10x Visium 小鼠脑数据演示最常用的一条流程:读入 → 质控 → 空间感知标准化(SCT)→ 聚类 → 空间变异基因 → 与参考 scRNA-seq 数据做 cell type 反卷积。
空间数据 vs 单细胞数据:换的是什么
很多新手把空间数据当作"加了坐标的单细胞"。这是错的,会带来三个直接后果:
| 维度 | scRNA-seq | Visium |
|---|---|---|
| 一个观测单元 | 1 个细胞 | 1 个 spot ≈ 1-10 个细胞混合 |
| 细胞类型注释 | 直接给 cluster 命名 | 要做反卷积估比例,单一 cell type 标签会误导 |
| 差异分析 | 比较两群细胞 | 比较两个空间区域,要考虑组织结构混杂 |
| 主问题 | "这是什么细胞" | "这块组织在干什么" |
Visium 的核心价值是空间结构,不是细胞分辨率。如果只想做精细的 cell type 分型,scRNA-seq 更合适;想问"哪些区域的免疫浸润强"、"肿瘤边界在分泌什么"、"白质和灰质的界面是什么",才轮到空间技术。
更高分辨率技术(Xenium、MERFISH)在单细胞分辨率,但牺牲了通量(可测的基因数 200-1000 个,远少于 Visium 的全转录组)。没有"分辨率高、通量也高"的免费午餐,先想清楚问题再选技术。
主流空间技术对比
| 技术 | 分辨率 | 数据量 | 常用工具 |
|---|---|---|---|
| 10x Visium | spot(~55 μm) | 中等 | Seurat、Scanpy + Squidpy |
| Slide-seq V2 | ~10 μm | 中等 | Seurat、RCTD |
| Stereo-seq | 亚微米 | 大 | StereoPy |
| MERFISH / seqFISH+ | 单细胞 | 大 | Squidpy、Giotto |
| Xenium | 亚细胞 | 大 | 10x pipeline、Seurat |
Visium 仍然是门槛最低的选择:一张切片产出的 spot 数在几千级,普通笔记本能直接分析,教材、社区工具链也最完整。
用 Seurat 分析 Visium 小鼠脑
从 10x Genomics 下载 Visium 小鼠脑切片数据后(或用 SeuratData 里的 stxBrain),基本流程和 scRNA-seq 很像,只是多了 spatial 这一层坐标和组织图片:
library(Seurat)
# 若要一键获取示例数据:
# InstallData("stxBrain")
# brain <- LoadData("stxBrain", type = "anterior1")
# 或者从本地目录读取
brain <- Load10X_Spatial(
data.dir = "~/biof3-data/visium-mouse-brain/outs",
filename = "filtered_feature_bc_matrix.h5",
assay = "Spatial",
slice = "slice1"
)
# 质控:在组织图上看每个 spot 的总表达量
VlnPlot(brain, features = "nCount_Spatial", pt.size = 0.1) + NoLegend()
SpatialFeaturePlot(brain, features = "nCount_Spatial")
两张 QC 图放一起看:有没有"测序偏低的区域"集中在切片边缘(组织脱落),或者局部异常高(气泡、污染)。
标准化这一步建议用 SCTransform,它对 spot 间差异较大的空间数据表现稳定:
brain <- SCTransform(brain, assay = "Spatial", verbose = FALSE)
brain <- RunPCA(brain, assay = "SCT")
brain <- FindNeighbors(brain, reduction = "pca", dims = 1:30)
brain <- FindClusters(brain, verbose = FALSE)
brain <- RunUMAP(brain, reduction = "pca", dims = 1:30)
# 聚类结果同时画在 UMAP 和组织切片上
DimPlot(brain, label = TRUE)
SpatialDimPlot(brain, label = TRUE, label.size = 3)
小鼠脑 Visium 在 SpatialDimPlot 里会呈现皮层、海马、胼胝体等解剖结构对应的聚类,不做任何 cell type 注释也能看出大致分区。
空间变异基因
常规的 FindVariableFeatures 只考虑表达变异,不考虑位置。空间变异基因是"表达分布不是随机的",通常也是真正有解剖学意义的基因:
brain <- FindSpatiallyVariableFeatures(
brain,
assay = "SCT",
features = VariableFeatures(brain)[1:1000],
selection.method = "moransi"
)
top_features <- head(
SpatiallyVariableFeatures(brain, selection.method = "moransi"),
6
)
SpatialFeaturePlot(brain, features = top_features, ncol = 3, alpha = c(0.1, 1))
"moransi"(Moran's I)衡量的是邻近 spot 的表达相似度,值越高说明越空间聚集。alpha 是作图参数,用来突出高表达区域、弱化背景。
真实示例:Visium 小鼠脑 Anterior 切片
配套脚本 module10_spatial_sci.R 用 SeuratData::stxBrain 的 anterior1 切片(10x Visium 小鼠脑前部,约 2700 个 spot)把上面的流程跑完。首次运行会通过 InstallData("stxBrain") 把数据装到本地(~136 MB),之后每次复用不再下载。
Rscript scripts/single-cell/sc10_spatial_sci.R
脚本顺序:LoadData 加载 Seurat 对象 → 在组织图上画 QC → SCTransform 标准化 → PCA / Leiden 聚类 / UMAP → 在 UMAP 和切片上各看一遍 cluster → 画四个已知解剖学 marker 的空间表达 → 用 Moran's I 找空间变异基因 → 单基因在 UMAP 和切片上的同步视图。
每张图看什么
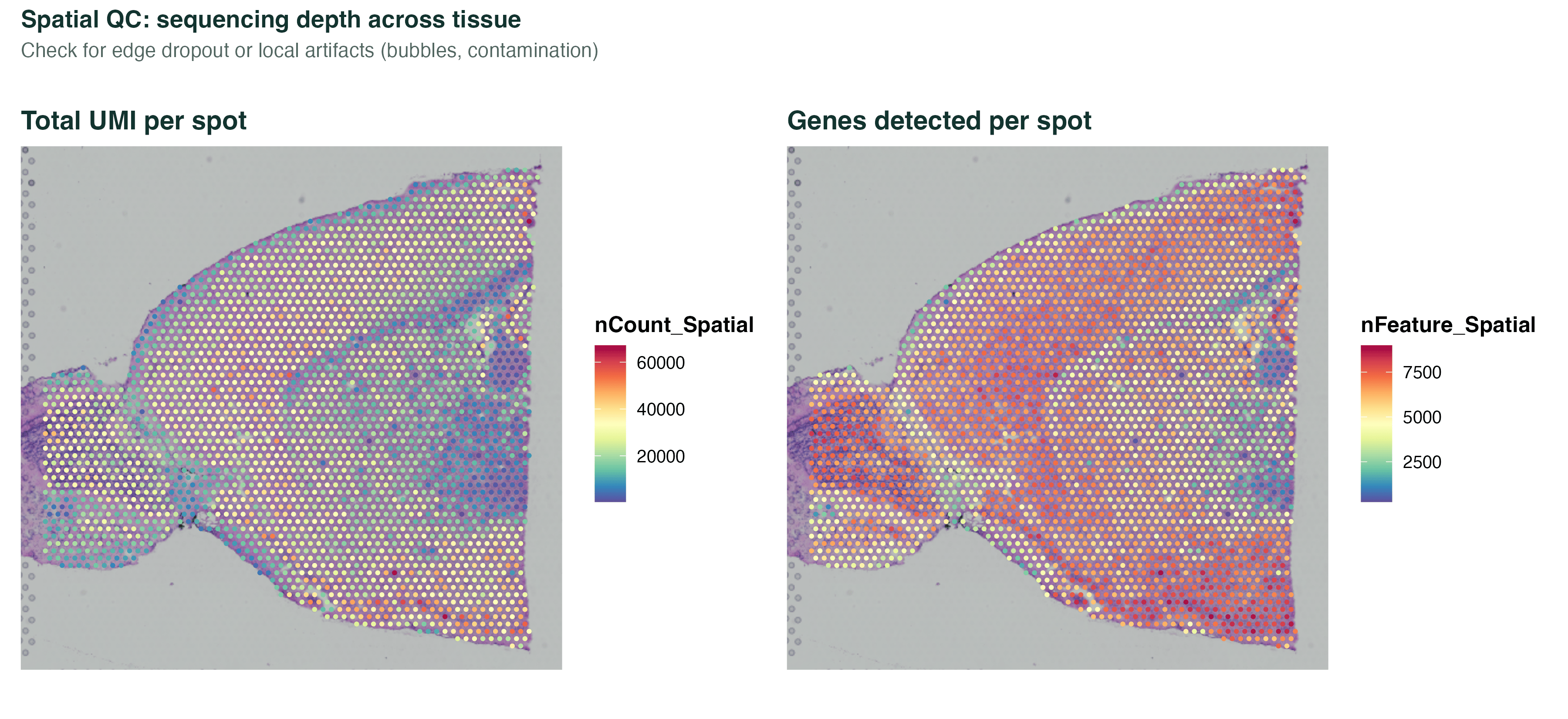
图 1:左图是每个 spot 的总 UMI 数(测序深度),右图是检测到的基因数。切片边缘深度偏低是正常的(组织少),内部如果出现一块局部异常高的亮斑就要警惕气泡、折叠或污染。这一步直接看切片比看小提琴图更直观。
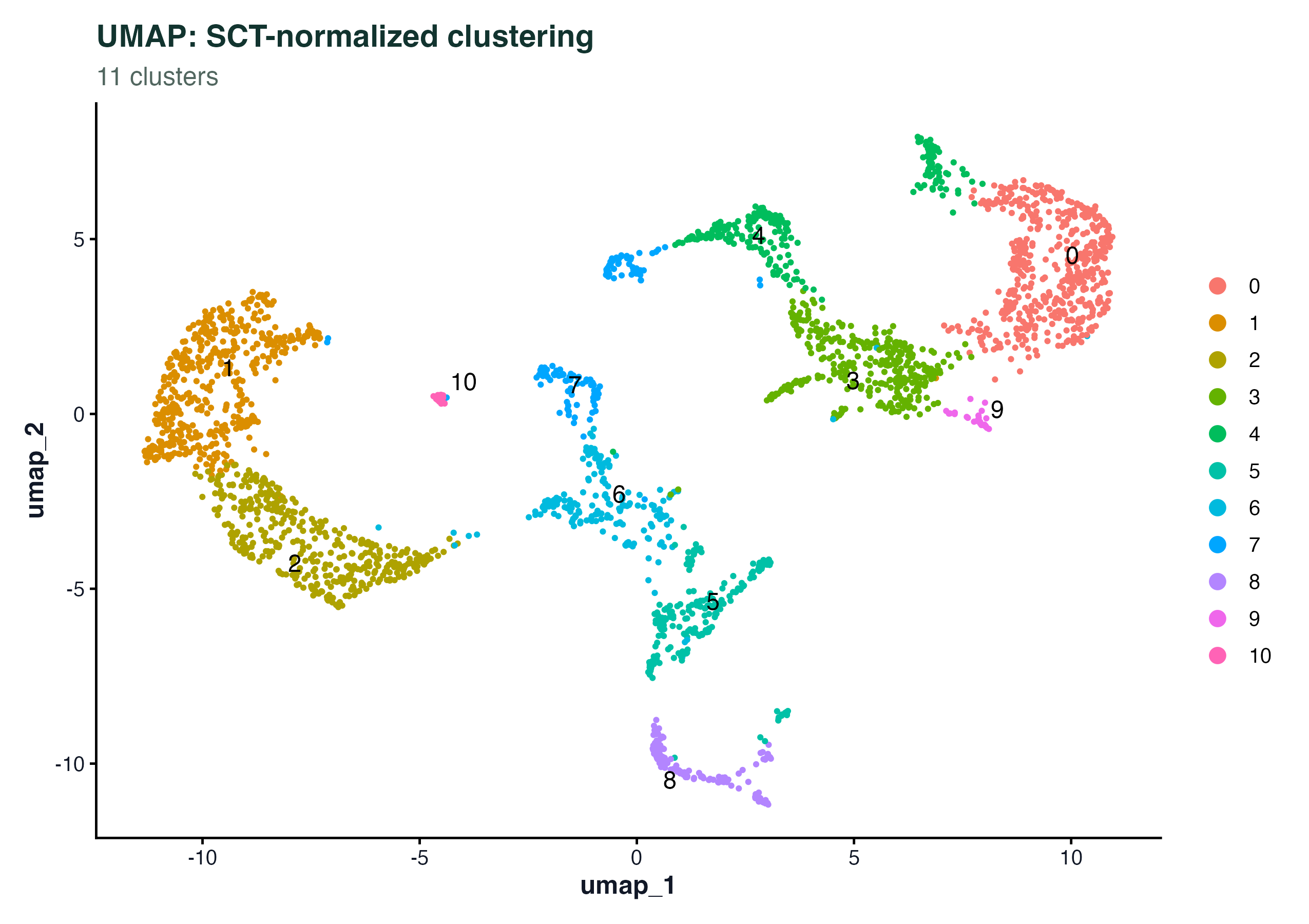
图 2:SCT 归一化后在 PCA 上跑 Leiden 得到的聚类,投到 UMAP 上。和 scRNA-seq 不同,Visium 的 cluster 数量不会太多(10 几个),因为一个 spot 代表的是几个细胞的混合。

图 3:同一个 seurat_clusters 直接画到组织切片上。不用做任何 cell type 注释,已经能看到皮层(同心环层)、海马(马蹄形)、胼胝体(中央带状)、脉络丛(点状)自然对应上不同的 cluster。这是 Visium 最"震撼"的一张图:空间聚类 = 解剖学结构。
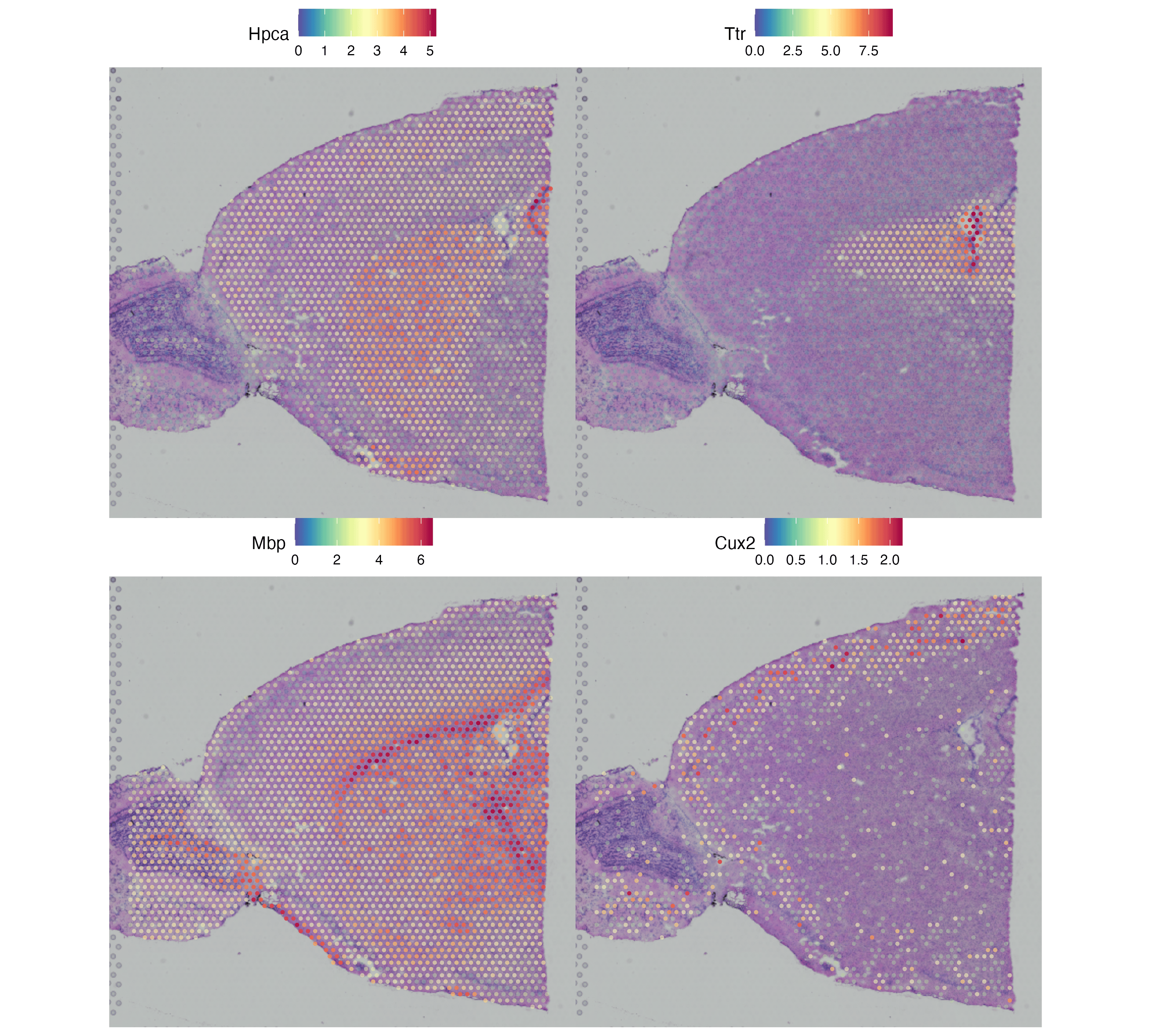
图 4:四个经典小鼠脑 marker 的空间表达 —— Hpca(海马)、Ttr(脉络丛)、Mbp(胼胝体 / 白质)、Cux2(皮层 II/III 层)。把这张图和图 3 一起看,能验证上一步的聚类是否确实落到了对应的解剖结构上。真实项目里换上自己关心的基因就能直接读出组织定位。
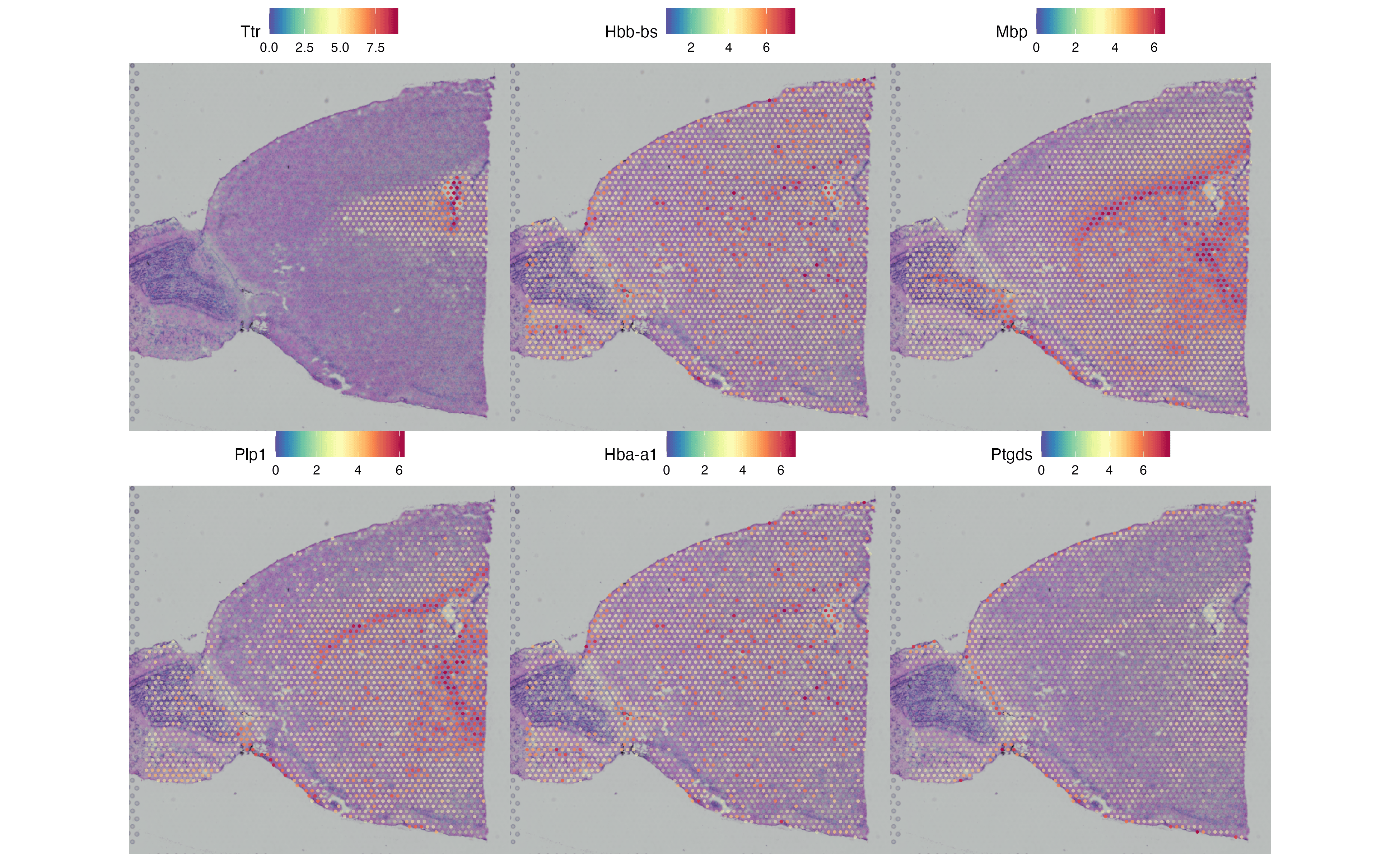
图 5:用 Moran's I 在前 1000 个高变基因里排名,取 Top 6 画空间分布。脚本实际跑出来是 Ttr, Hbb-bs, Mbp, Plp1, Hba-a1, Ptgds —— 脉络丛、红细胞、胼胝体、少突胶质、红细胞、脑膜/神经干细胞 —— 每一个都对应清晰的解剖结构,而不是随机散布,这正是空间变异基因要挑的信号。
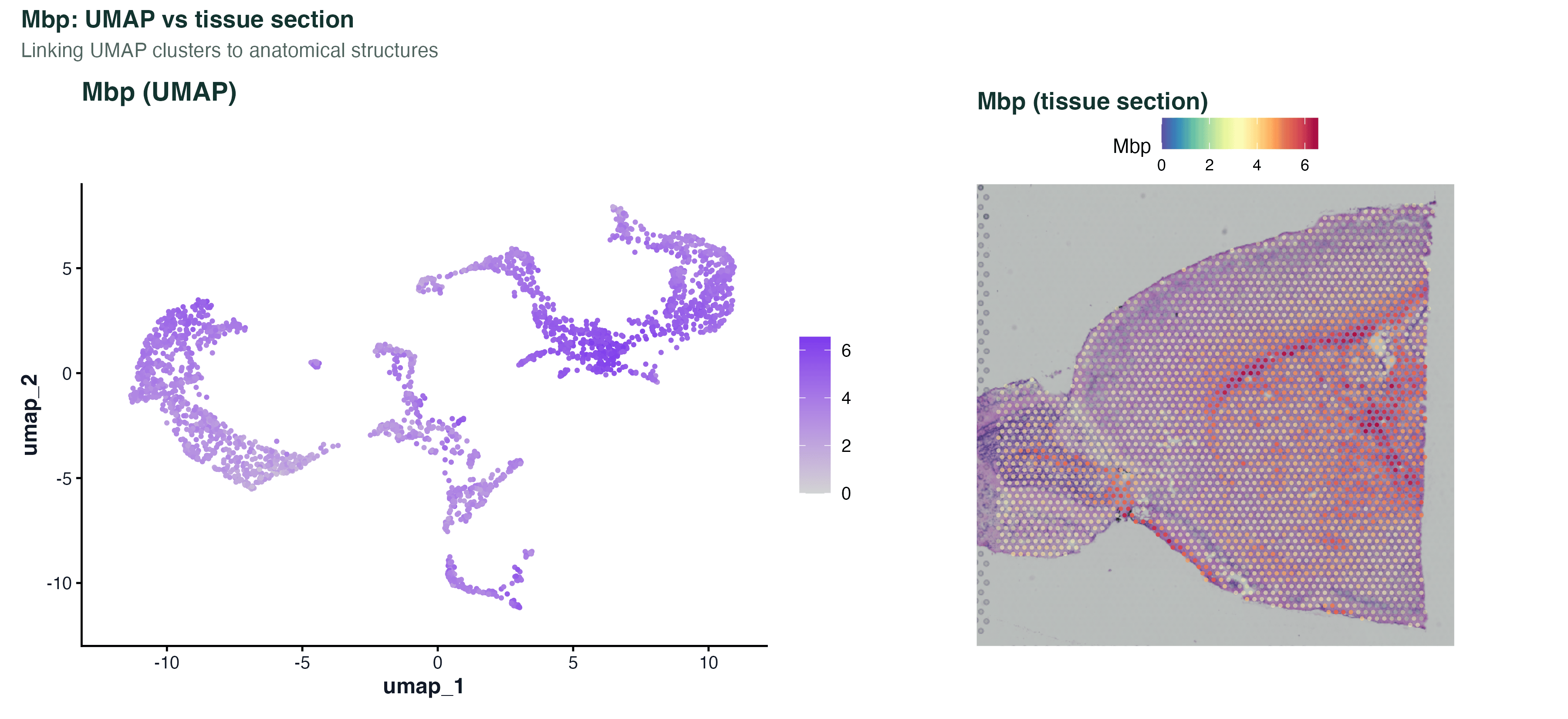
图 6:把 Mbp 在 UMAP 和组织切片上各画一张放一起。左边看它对应 UMAP 里的哪一个 cluster,右边看它对应切片上的什么结构。当一个未知基因的 UMAP 分布集中但你又不知道它的生物学意义时,拿它跟切片版一起看,经常能马上定位到"这是 XX 层 / XX 核团的 marker"。
套到自己数据上
脚本对小鼠脑做了演示。自己的切片(人脑、肿瘤、肝、皮肤等)换 LoadData 那行为 Load10X_Spatial("~/path/to/outs"),其余不动就行。marker_genes 按组织换:比如做肿瘤切片,可以换成 EPCAM(上皮)、PTPRC(免疫)、COL1A1(基质�);做肝切片可以换成 ALB(肝细胞)、GLUL(周中静脉)、CYP2E1(三区带)。分辨率更高的技术(Stereo-seq、Xenium)数据量更大,配合 Python 的 Squidpy 更流畅。
把 scRNA-seq 当作参考做反卷积
Visium 的每个 spot 通常包含多个细胞,直接做 cell type 注释会模糊。常用做法是把已经注释好的 scRNA-seq 数据当参考,把 spot 内的细胞组成估出来。常用工具有 RCTD、SPOTlight、CARD:
# SPOTlight 示例(输入:scRNA 参考 + Visium 对象)
library(SPOTlight)
sc_ref <- readRDS("pbmc_reference.rds") # 假设已分析并注释
markers <- Seurat::FindAllMarkers(sc_ref)
spotlight_res <- SPOTlight(
x = sc_ref,
y = brain,
groups = sc_ref$cell_type,
mgs = markers
)
plotSpatialScatterpie(
x = brain,
y = spotlight_res,
cell_types = unique(sc_ref$cell_type),
img = FALSE
)
得到的 scatterpie 在每个 spot 位置画一个小饼图,显示各 cell type 的估计比例。常见验证方式:查看某种 cell type 的比例在组织切片上是否和已知解剖学位置一致。
用 Scanpy + Squidpy 做 Python 版
Python 生态下 scanpy 配合 squidpy 可以覆盖类似流程,并提供更丰富的空间统计(nhood enrichment、co-occurrence 等):
import scanpy as sc
import squidpy as sq
adata = sc.read_visium("~/biof3-data/visium-mouse-brain/outs")
adata.var_names_make_unique()
sc.pp.calculate_qc_metrics(adata, inplace=True)
sc.pl.spatial(adata, color="total_counts")
sc.pp.normalize_total(adata, inplace=True)
sc.pp.log1p(adata)
sc.pp.highly_variable_genes(adata, flavor="seurat", n_top_genes=2000)
sc.pp.pca(adata)
sc.pp.neighbors(adata)
sc.tl.umap(adata)
sc.tl.leiden(adata)
sc.pl.spatial(adata, color="leiden")
# 空间邻域富集:看哪些 cluster 在空间上彼此相邻
sq.gr.spatial_neighbors(adata)
sq.gr.nhood_enrichment(adata, cluster_key="leiden")
sq.pl.nhood_enrichment(adata, cluster_key="leiden")
Scanpy/Squidpy 侧最适合的场景是高分辨率空间数据(Stereo-seq、MERFISH),它们对大矩阵更友好。Visium 级别数据两套都能跑,选择��看个人生态。
常见坑
坑 1:只下了 filtered_feature_bc_matrix,没下 spatial 文件夹
Visium 的"空间"信息(图片 + spot 坐标)放在 spatial/ 子目录,不在表达矩阵 tar 包里。下载时要同时下 _spatial.tar.gz,否则 Load10X_Spatial 会报错或画不出切片图。这个坑在 01 章 PBMC 部分已经提过,做空间项目时再重提一次。
坑 2:用 scRNA-seq 那套阈值过滤 spot
nFeature_RNA < 200 在单细胞合理,但 spot 是几个细胞的混合,正常 spot 通常 > 1000 个基因。组织薄、深度低的边缘 spot 可能 < 500,但要看是组织本身就稀疏(脑的白质区域基因表达本来就少)还是切片缺陷。先 SpatialFeaturePlot 看分布,再定阈值,不要直接套单细胞经验值。
坑 3:把 Visium spot 当单细胞做差异表达
每个 spot 是 1-10 个细胞的混合,spot 间差异 = 细胞类型组成差异 + 表达差异混在一起。直接 FindMarkers 出来的差异基因没法解释成"该细胞类型上调了 X" — 可能只是这个区域多了某类细胞。需要先做反卷积估比例,再做基于比例的统计建模。
坑 4:Moran's I 选高变基因后跑,把组织背景都筛掉了
FindSpatiallyVariableFeatures 默认只在前 1000 个高变基因里搜,有些有解剖学意义的基因(脑里的 Mbp、Plp1)虽然空间高度聚集但不是 top 高变基因,会被漏掉。要么扩大候选范围(features = VariableFeatures(brain)[1:5000]),要么直接用全基因,代价是慢一些。
坑 5:反卷积参考集和 Visium 数据物种 / 状态不匹配
用人 PBMC scRNA-seq 当参考,去反卷积小鼠脑 Visium — 直接错。反卷积参考集必须和目标数据物种一致、组织来源接近。理想情况是同实验室同批次做的 scRNA-seq 当参考,至少也要是同物种同组织的公开数据。
在线分析:不想配环境,直接在 BioF3 跑
上面这套流程要本地装 Seurat / Squidpy / 反卷积包,对新手不友好。BioF3 把空间转录组的核心分析做成了云平台 + 在线工具两条线,浏览器里就能跑,后端用 scanpy + Squidpy(Python 生态,空间分析比 R 更全)。
空间组学云平台(/spatial)
空间组学云平台 和单细胞云平台操作习惯完全一致:新建项目 → 上传 .h5ad 数据集 → 选分析类型 → 看结果 + AI 解读。适合要管理多个项目 / 多次分析串起来的场景。当前支持 5 类分析:标准流程、重聚类、空间高变基因、细胞解卷积、配体受体。
上传要求:
.h5ad(AnnData)含obsm['spatial']坐标。10x Visium 标准输出直接可用。
在线工具(/tools,单次分析不建项目)
如果只想跑一次、不想建项目,用 /tools 下的空间组学工具,上传文件一键出 SCI 报告:
| 工具 | 对应本章内容 | 入口 |
|---|---|---|
| 空间转录组标准分析 | QC → 聚类 → UMAP → 邻域富集 → SVG(本章主线流程) | /tools/#spatial-standard |
| 空间高变基因(SVG) | Moran's I + Geary's C + Sepal 三方法共识(比本章单一 Moran's I 更稳) | /tools/#spatial-svg |
| 空间细胞解卷积 | Tangram 把单细胞参考映射到 spot(对应本章反卷积小节) | /tools/#spatial-deconv |
| 空间配体受体 | LIANA 空间感知 LR(本章没展开,是空间通讯的进阶玩法) | /tools/#spatial-lr |
每个工具都带真实 Visium 小鼠脑公开 demo(进工具页点"报告"按钮即可看),不上传也能先看效果。
想自己改图? 这些工具的输出图都能在 FigCode 找到对应卡片(
spatial-cluster/feature-spatial/spatial-svg-moran/spatial-deconv-c2l/spatial-lr-liana/squidpy-neighbors),复制代码改配色 / 改基因即可。
下载资源
下一步
接着深入:
- 10 scATAC-seq 分析 — 单细胞专题的最后一篇,染色质层面的分析
- 06 细胞-细胞通讯分析 — 在空间数据上做通讯分析比 scRNA-seq 可靠得多,因为能直接验证 sender 和 receiver 的物理距离
横向延伸:
- 03 质量控制、聚类与细胞类型注释 — 反卷积参考集就是用 03 章流程注释好的 scRNA-seq 数据
- Squidpy 文档 — Python 端做空间统计(邻域富集、共定位)的标准工具
参考资源
离线资料下载
手册 HTML / PDF 已在后台预生成,点击后直接下载网站静态资源。