05 轨迹推断与拟时序分析
聚类把细胞分成若干离散的 cluster,但发育、分化、应激响应这类现象更自然的描述是"细胞沿着一条连续路径逐渐变化"。轨迹推断(trajectory inference)的目标就是把单细胞数据里的连续结构显式地建模成图或曲线,然后给每个细胞一个在这条路径上的位置 —— 就是拟时序(pseudotime)。
这是一个"真正的时间轴"在单细胞里其实不可观测的近似:所有细胞都是同一刻被测的,我们只是用基因表达的变化推测出它们在分化进程里前后的相对位置。本章走三种主流方法的典型用法:Monocle3、Slingshot、以及基于 RNA velocity 的 scVelo。
这类分析能回答什么
- 分化路径上关键的基因开关在哪个拟时点打开
- 某个 cluster 在分化树上处在分支点还是端点
- 两个最终命运(比如 T vs B)的分叉什么时候发��生
- 哪些基因是"驱动基因"、哪些只是被动跟随
如果你的项目里没有明显的连续过程(比如只是健康样本的静态图谱),拟时序分析往往得不出有用的结论。先想清楚要回答什么问题再选方法。
先确认你的数据该不该做轨迹
不是所有数据都适合做轨迹。先问自己三个问题:
1. 系统里真有连续过程吗?
适合:发育(胚胎、造血)、分化(干细胞 → 成熟细胞)、应激响应(药物处理时间序列)、肿瘤进展。
不适合:健康成人的稳态组织(PBMC、肾、肝),cluster 之间多数是平行关系而不是前后关系。强行做出来的"轨迹"是数学拟合的副产品,不反映生物学。
2. 起点选得出来吗?
任何拟时序方法都需要一个起点,错的起点出错的轨迹。判断标准:
- 有早期分化 marker(干细胞标志:CD34、PROM1、SOX2 等)
- 有 RNA velocity(spliced/unspliced 比例能看出方向)
- 有外部时间信息(Day0 / Day3 / Day7 这种采样设计)
三者都没有时,做轨迹基本是凭经验在猜。
3. 数据深度够吗?
拟时序对 dropout 特别敏感。每个细胞 nFeature_RNA < 1000 时,分支点判断会很不稳。深度不够时先增加测序深度再做,不要硬上。
方法选择
| 方法 | 语言 | 擅长 | 备注 |
|---|---|---|---|
| Monocle3 | R | 复杂分支结构 | 现在单细胞轨迹分析的默认选择 |
| Slingshot | R | 简单线性 / 少分支 | 需要先聚类,但接口简单 |
| scVelo | Python | 方向性强的分化路径 | 需要 spliced / unspliced counts,通常从 velocyto 或 Cell Ranger 输出生成 |
| PAGA | Python (Scanpy) | 大规模数据的拓扑结构 | 抽象程度高,适合做总览图 |
| CytoTRACE | R | 估计每个细胞的分化程度 | 不给出显式轨迹,只给出相对排序 |
实际选择建议:
- 新项目从 Monocle3 开始。它是现在最标准的选择。
- 需要 RNA velocity 的"方向性"(看细胞正在往哪变)时用 scVelo,前提是你有 BAM 文件或能跑 velocyto。
- 只是想给细胞一个"早-晚"排序、不关心轨迹形状的话 CytoTRACE 最简单。
Monocle3:完整流程
安装
if (!requireNamespace("BiocManager", quietly = TRUE)) install.packages("BiocManager")
BiocManager::install(c(
"BiocGenerics", "DelayedArray", "DelayedMatrixStats", "limma",
"S4Vectors", "SingleCellExperiment", "SummarizedExperiment",
"batchelor", "HDF5Array", "terra", "ggrastr"
))
install.packages("devtools")
devtools::install_github("cole-trapnell-lab/monocle3")
从 Seurat 对象出发
Monocle3 的核心对象叫 cell_data_set(CDS)。如果前面用 Seurat 分析过,可以直接转:
library(Seurat)
library(monocle3)
library(SeuratWrappers)
cds <- as.cell_data_set(seurat_obj)
也可以手动从 counts 矩阵建:
cds <- new_cell_data_set(
expression_data = seurat_obj@assays$RNA@counts,
cell_metadata = seurat_obj@meta.data,
gene_metadata = data.frame(
gene_short_name = rownames(seurat_obj),
row.names = rownames(seurat_obj)
)
)
预处理、降维和聚类
cds <- preprocess_cds(cds, num_dim = 50)
cds <- reduce_dimension(cds, reduction_method = "UMAP")
cds <- cluster_cells(cds, resolution = 1e-3)
plot_cells(cds, color_cells_by = "cell_type")
plot_cells(cds, color_cells_by = "cluster")
Monocle3 自己有一套 UMAP 和聚类。如果你已经在 Seurat 里做过一套,结果通常很相似,但 Monocle3 的 UMAP 会是后续 learn_graph 的输入,不要跳过��这一步。
学习轨迹图
cds <- learn_graph(cds)
plot_cells(cds,
color_cells_by = "cell_type",
label_groups_by_cluster = FALSE,
label_leaves = FALSE,
label_branch_points = FALSE
)
learn_graph 会在 UMAP 上拟合一条主干曲线(principal graph),分支点和叶子结点由算法自动判断。
选择起点并排序
拟时序需要一个起点("分化程度最低的那个 cluster")。两种方式:
# 方式 1:点图里手动点
cds <- order_cells(cds)
# 方式 2:根据已有注释自动选
cds <- order_cells(cds, root_cells = colnames(cds)[cds$cell_type == "Stem"])
plot_cells(cds,
color_cells_by = "pseudotime",
label_cell_groups = FALSE,
label_leaves = FALSE,
label_branch_points = FALSE
)
找随拟时序变化的基因
track_genes <- graph_test(cds, neighbor_graph = "principal_graph")
track_genes_sig <- track_genes |>
dplyr::filter(q_value < 0.05) |>
dplyr::arrange(q_value)
head(track_genes_sig, 20)
plot_cells(
cds,
genes = head(track_genes_sig$gene_short_name, 4),
show_trajectory_graph = FALSE,
label_cell_groups = FALSE
)
基因模块
把这些随拟时序变化的基因聚成共表达模块,能看清分化不同阶段各自"开启"了哪一组程序:
gene_modules <- find_gene_modules(
cds[track_genes_sig$gene_short_name, ],
resolution = 1e-2
)
plot_cells(
cds,
genes = gene_modules,
label_cell_groups = FALSE,
show_trajectory_graph = FALSE
)
Slingshot:轻量替代
分支结构简单、已经聚好类的情况下,Slingshot 更直接:
BiocManager::install("slingshot")
library(slingshot)
library(SingleCellExperiment)
sce <- as.SingleCellExperiment(seurat_obj)
sce <- slingshot(
sce,
clusterLabels = "seurat_clusters",
reducedDim = "UMAP",
start.clus = "0"
)
pseudotime <- slingPseudotime(sce)
head(pseudotime)
输出的 pseudotime 是一个 细胞 × 分支数 的矩阵,每列对应一条分支的拟时序值。可视化:
library(RColorBrewer)
colors <- colorRampPalette(brewer.pal(11, "Spectral")[-6])(100)
plot(
reducedDims(sce)$UMAP,
col = colors[cut(pseudotime[, 1], breaks = 100)],
pch = 16,
asp = 1
)
lines(SlingshotDataSet(sce), lwd = 2, col = "black")
找随拟时序变化的基因走 tradeSeq:
library(tradeSeq)
sce <- fitGAM(sce)
assocRes <- associationTest(sce)
sig_genes <- rownames(assocRes)[assocRes$pvalue < 0.05]
真实示例:在 PBMC 3k 上跑一次 Slingshot
PBMC 3k 不是典型的分化数据(它是健康外周血的几种稳定免疫细胞),但 Slingshot 在它上面能拟出一条合理的主曲线,用来演示"读图"的过程绰绰有余。下面的六张图全部来自配套脚本 module06_trajectory_sci.R 在真实 PBMC 3k 矩阵上跑出的结果,跑一次大约 1 分钟:
Rscript scripts/single-cell/sc06_trajectory_sci.R
脚本会下载 PBMC 3k、做完 QC/归一化/PCA/UMAP/聚类、按经典 marker 给每个细胞一个粗注释(B、T CD4、T CD8、NK、Monocyte、DC、Platelet),然后用 Slingshot 从 Monocyte 起点建主曲线、拿到拟时序、把结果画出来。
每张图看什么
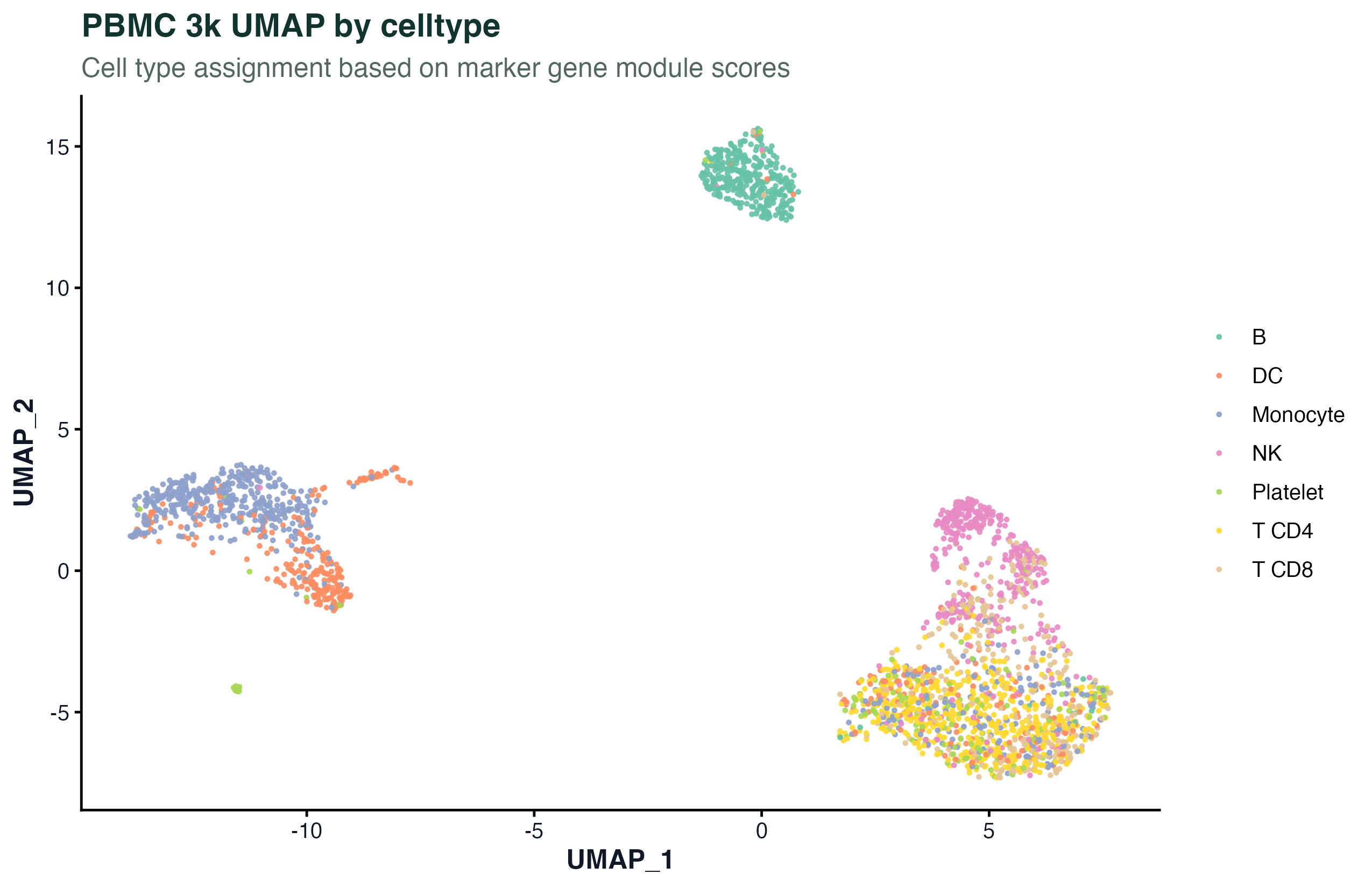
图 1:PBMC 3k 的 UMAP,按经典 marker 给出的粗注释上色。这张图不是轨迹推断的产物,只是让后面的结果有参照。
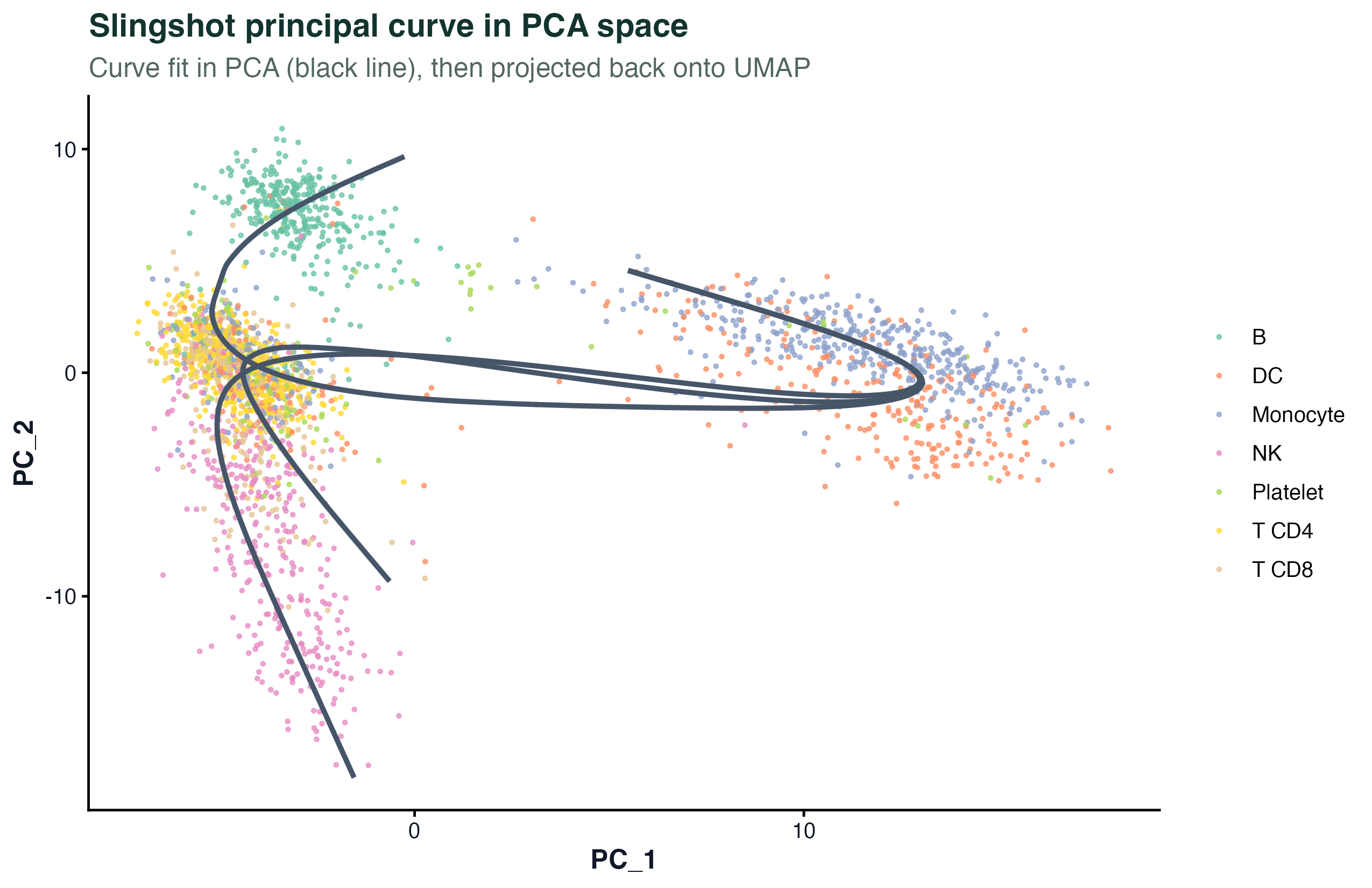
图 2:Slingshot 在 PCA 空间拟合的主曲线(实线)。每条 lineage 对应一条分支,PBMC 3k 拟出一两条分支很常见——免疫系统里细胞之间关系不是严格的线性分化,所以曲线形状不要解读成"真实发生了这条路径的分化"。
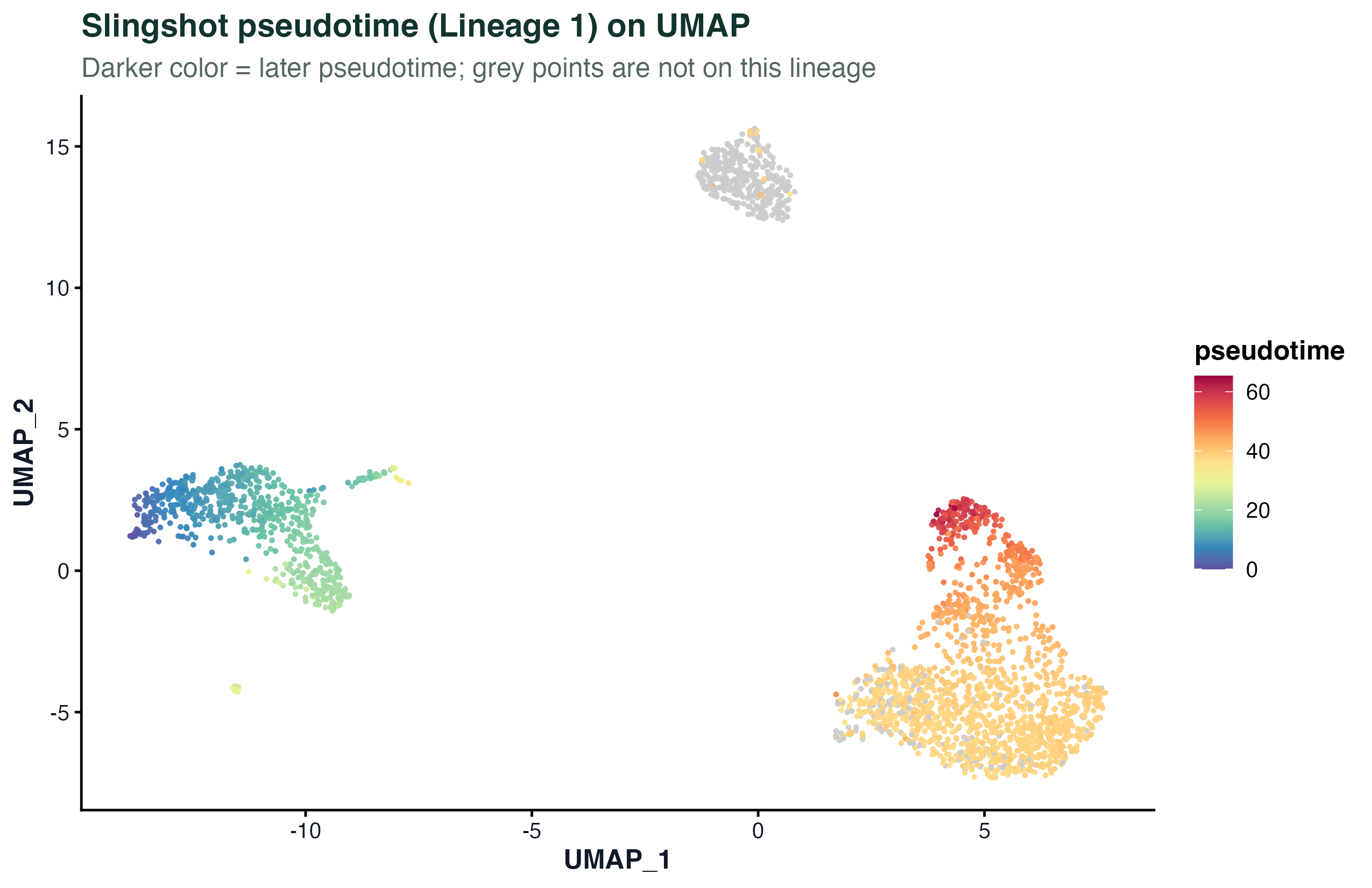
图 3:主分支(Lineage 1)的拟时序值投到 UMAP 上。颜色越深的细胞在这条分支上越"晚"。灰色点是不被这条分支覆盖的细胞。
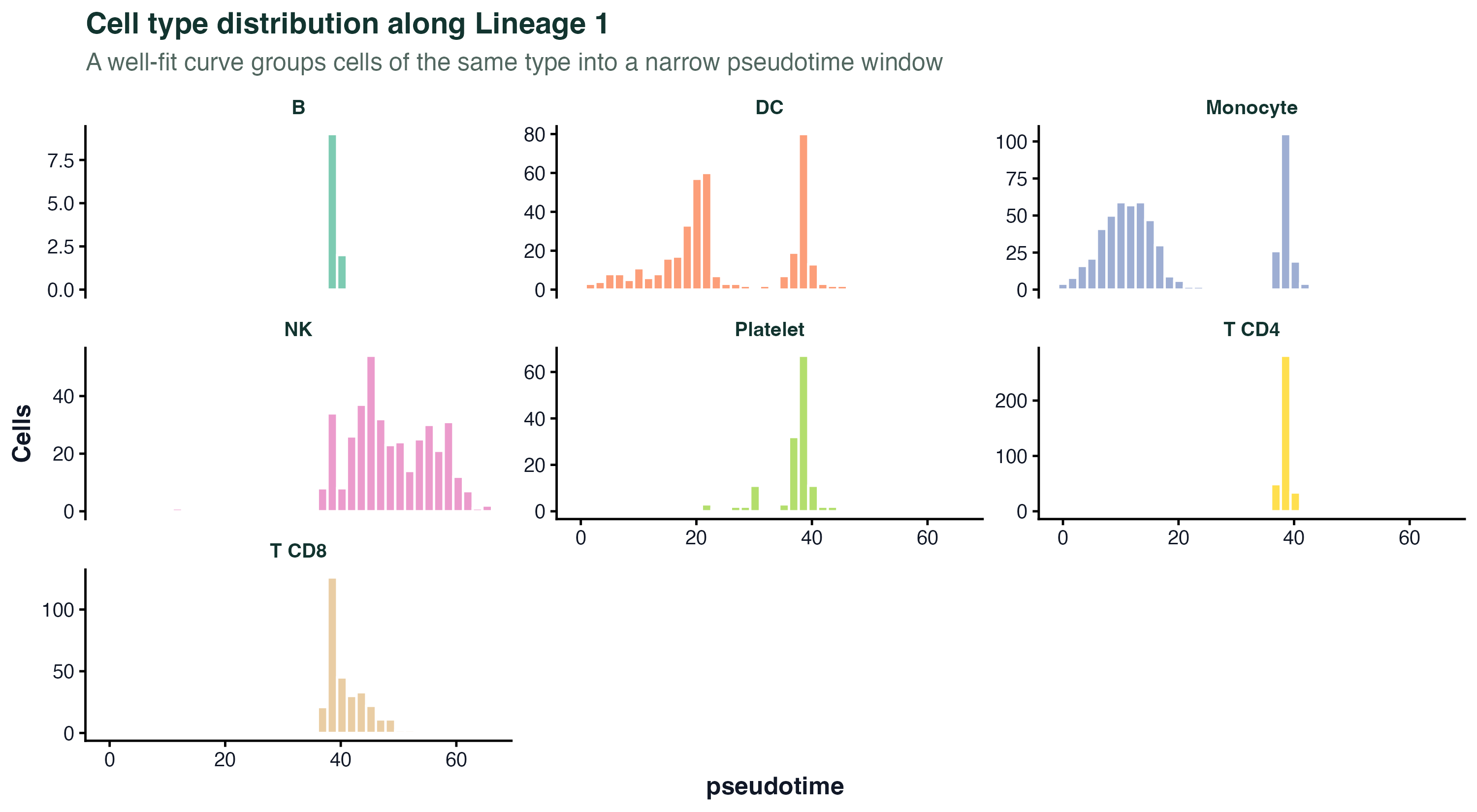
图 4:每类细胞的拟时序分布。如果主曲线合理,同一 celltype 的细胞应该在某个区段聚集。这张图是判断"起点 / 方向"是否设对的第一扇窗。
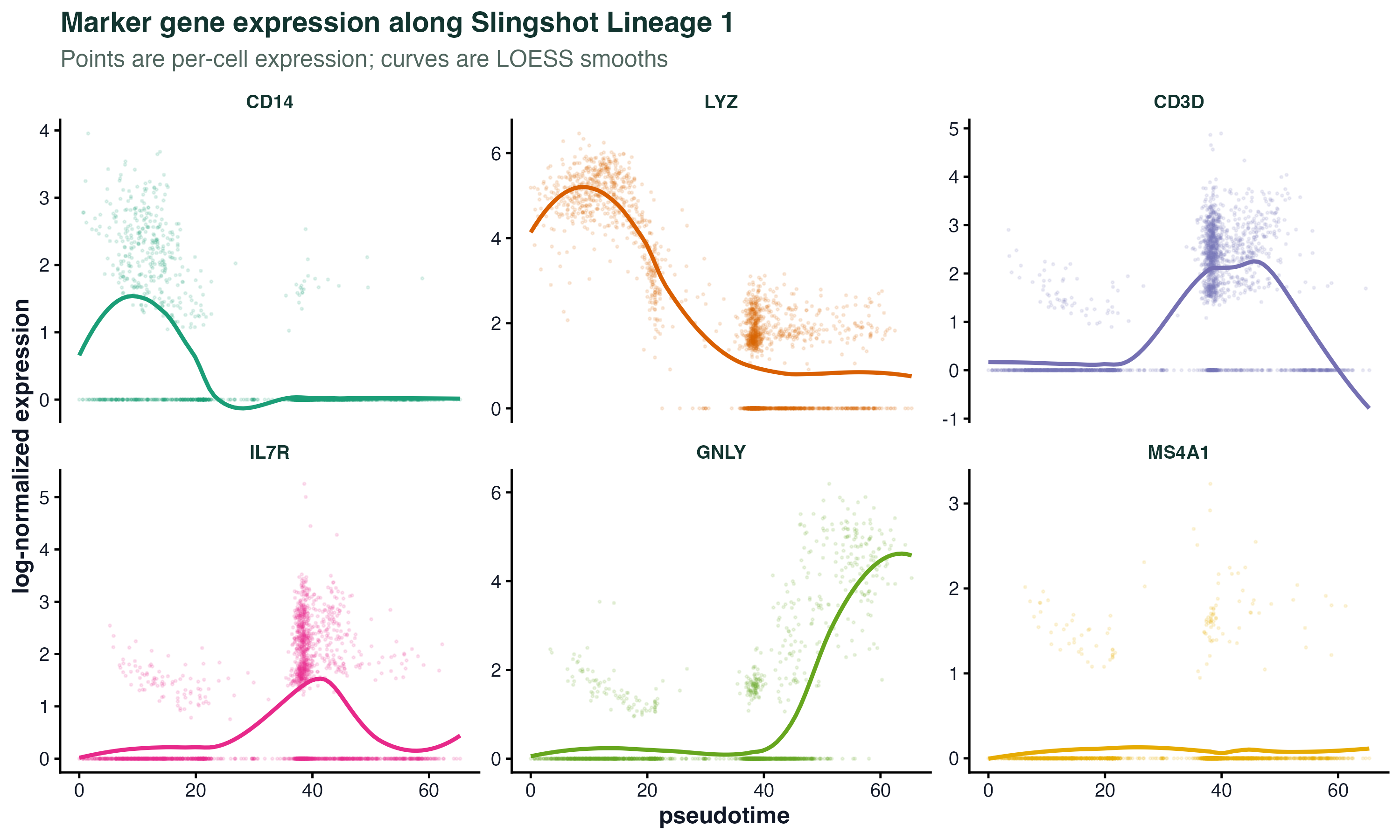
图 5:六个经典 marker 沿拟时序的表达曲线(散点是每细胞值,曲线是 LOESS 平滑)。CD14/LYZ 在前段高、CD3D/IL7R 在中段高、GNLY/MS4A1 往后段倾斜——这是一份典型的"免疫谱系变化剪影"。
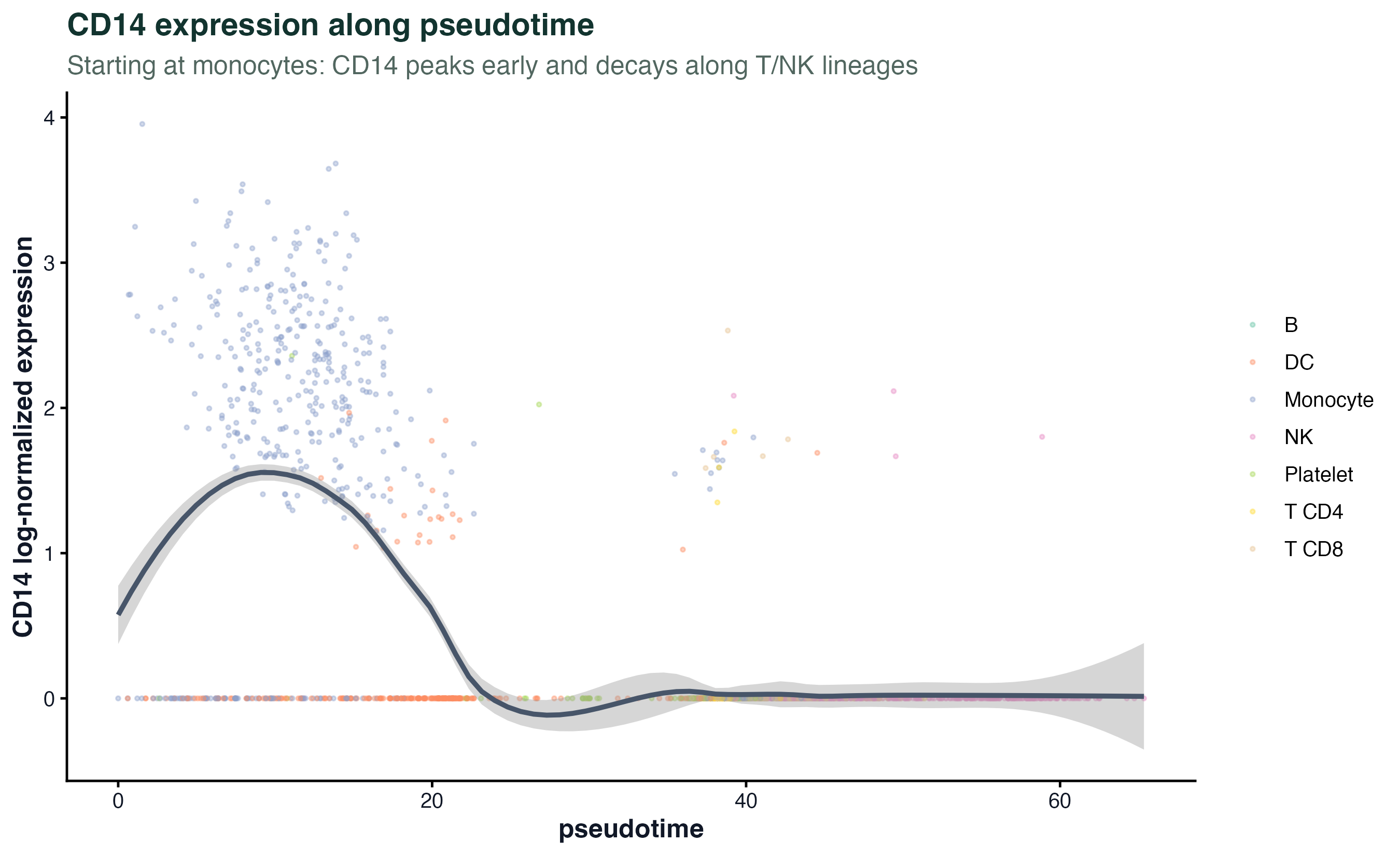
图 6:单独把 CD14 拎出来看。这种"单基因沿拟时序"图是展示结果时最简洁的版本,论文里常用。
想把这套流程套到自己的数据上
把第 35-45 行的读数据部分换成自己的 Read10X() 或 readRDS(),再改动 marker_signature 列表和 start.clus,剩下的代码基本不用动。真实分化数据(造血、胚胎发育、CD8 记忆-效应)在这套流程下会给出更有说服力的轨迹。
scVelo:用 RNA velocity 给出方向
标准拟时序只回答"这些细胞的相对位置",但不回答"他们正在往哪变"。RNA velocity 利用未剪接 mRNA 和已剪接 mRNA 的比例估计每个细胞的"即时变化方向",在分化终点还没进入数据的项目里特别有用。
前提:有包含 spliced / unspliced counts 的 loom 文件。通常走 velocyto 生成:
velocyto run10x -m repeat_msk.gtf /path/to/sample01 genes.gtf
Python 分析:
import scanpy as sc
import scvelo as scv
adata = scv.read("sample01.loom", cache=True)
adata_seurat = sc.read_h5ad("analyzed.h5ad")
adata = scv.utils.merge(adata, adata_seurat)
scv.pp.filter_and_normalize(adata, min_shared_counts=20, n_top_genes=2000)
scv.pp.moments(adata, n_pcs=30, n_neighbors=30)
scv.tl.velocity(adata)
scv.tl.velocity_graph(adata)
scv.pl.velocity_embedding_stream(adata, basis="umap", color="cell_type")
需要更精细的估计时用动态模型(dynamical mode),慢但准:
scv.tl.recover_dynamics(adata)
scv.tl.velocity(adata, mode="dynamical")
scv.tl.velocity_graph(adata)
scv.tl.velocity_pseudotime(adata)
scv.pl.scatter(adata, color="velocity_pseudotime", cmap="gnuplot")
PAGA 和 CytoTRACE:快速概览
PAGA 把聚类结构抽成一张图,每个 cluster 是节点,节点之间边的粗细代表"有多少细胞邻居跨 cluster":
sc.tl.paga(adata, groups="leiden")
sc.pl.paga(adata, color=["leiden", "CST3"])
sc.tl.draw_graph(adata, init_pos="paga")
sc.pl.draw_graph(adata, color="leiden", legend_loc="on data")
import numpy as np
adata.uns["iroot"] = np.flatnonzero(adata.obs["leiden"] == "0")[0]
sc.tl.dpt(adata)
sc.pl.umap(adata, color=["dpt_pseudotime"], color_map="viridis")
CytoTRACE 更简单,不建轨迹、只给每个细胞一个"分化程度估计值":
devtools::install_github("digitalcytometry/cytotrace")
library(CytoTRACE)
results <- CytoTRACE(as.matrix(seurat_obj@assays$RNA@counts))
seurat_obj$CytoTRACE <- results$CytoTRACE
FeaturePlot(seurat_obj, features = "CytoTRACE")
CytoTRACE 的假设是"分化程度高的细胞表达基因更少",对造血、上皮等几种经典系统比较可靠,放到任何系统上都用要谨慎。
结果解读的几个注意事项
- 拟时序值是相对的,不是真实时间。两个细胞 pt 差 2 和差 20 不代表真实时间差 10 倍。
- 起点错了整条轨迹都错。选 root 时如果没有分化早期的 marker,可以用 CytoTRACE 或 velocity 方向辅助判断。
- 分支点要交叉验证:Monocle3 的分支点和 marker 基因切换、已发表分化时序一致时才有说服力。
- 不要把轨迹当作因果。拟时序只表明"这些细胞在基因表达空间里排成了这样",转化成 "A 变成 B" 这个因果陈述需要额外证据(比如谱系追踪实验或 velocity 方向)。
常见坑
坑 1:起点是猜的
order_cells 弹出 GUI 让你点起点,很多人随便点一个或者直接挑中心位置。起点错了,下游所有结论都错。如果没有 CD34/PROM1 这种公认 marker,至少用 CytoTRACE 算一下分化得分,把得分最低的 cluster 当起点。
坑 2:把 PBMC 这类静态数据强行做轨迹
PBMC 是稳态外周血,T/B/NK/Mono 之间没有"先后"关系。Slingshot 会拟合出一条曲线,那��条曲线没有生物学意义。本章 PBMC 3k 示例只是演示读图方法,不要把"PBMC 3k 的轨迹"当生物学结论用。
坑 3:scVelo 没装好 velocyto 输出就跑
scVelo 需要 spliced/unspliced 两套矩阵,标准 Cell Ranger 输出没有这两套。要么用 velocyto run10x 重跑、要么用 starsolo + --soloFeatures Gene Velocyto。直接拿 filtered_feature_bc_matrix 喂 scVelo 是不行的。
坑 4:分支结构不稳定换种子就变
Monocle3 的 learn_graph 在边缘案例下对随机种子敏感。换 seed 跑 3-5 次,看主干是否一致。如果每次结果都很不一样,说明数据本身不支持这个轨迹结构 — 不要选其中"最顺眼"的一次发表。
坑 5:把"沿拟时序变化的基因"全堆图
graph_test 经常返回几千个显著基因,全画热图毫无信息量。先按生物学功能筛一遍:转录因子、信号通路成员、已知分化 marker,再画几十个最相关的。
下载资源
下一步
接着深入:
- 06 细胞-细胞通讯分析 — 把"沿轨迹变化的基因"放进配体-受体框架,看分化过程中哪些信号在驱动状态切换
横向延伸:
- 07 多模态数据分析 — 如果你有 ATAC 数据,染色质开放变化能给轨迹方向提供独立证据
- Saelens 2019 轨迹方法基准评测 — 系统对比了 45 种轨迹工具,决定方法时读这篇
参考资源
离线资料下载
手册 HTML / PDF 已在后台预生成,点击后直接下载网站静态资源。