module04
速答
Q: 单细胞整合什么时候需要做? A: 多样本 / 多批次时必须做。判断标准:同一细胞类型在不同样本的 UMAP 上分成两群 → 批次效应,需要整合。
Q: Seurat CCA / Harmony / scVI 怎么选? A: CCA 是 Seurat 经典方法,稳定但慢;Harmony 快且效果好,推荐首选;scVI 深度学习方法,数据量大(>50k 细胞)时优势明显。
Q: 整合过度会怎样? A: 把真实生物学差异也抹掉了(over-correction)。表现为 UMAP 上所有样本混成一团,看不到处理 vs 对照的差异。看整合后的 UMAP + 样本分布图判断。
Q: 整合后怎么验证成功? A: 同一细胞类型在 UMAP 上聚在一起(生物学信号保留),不同样本的同一类型混合(批次效应去除),但条件间的差异仍可见。
04 多样本数据整合
如果你手上只有一个样本,上一章的流程走完就能得到一份可用的注释。但真实项目几乎都有多个样本:治疗组对照组、几个个体、不同组织部位、不同时间点。把它们放在同一张 UMAP 上分析的时候,批次效应会让相同细胞类型在不同样本里显得像两群不同的细胞——这是任何方法上的差异都不能忽略的麻烦。
数据整合(integration)的目的就是把批次效应校正掉,让 "同一类细胞" 在多个样本之间对齐,而不把真实的生物学差异也一起抹掉。本章讲主流整合方法在什么场景下用,以及 Seurat / Harmony / scVI 的实际写法。
本页配图用的是 PBMC 3k 真实矩阵按 barcode 顺序切出的 subset,用来演示整合诊断图该怎么读。它不代表真实生物学批次,正式多样本项目应该把 subset 换成多个真实样本。
整合的核心问题:能去掉多少、不能去掉多少
整合不是越彻底越好。它在两件事之间做平衡:
- 消除批次效应:让同一类细胞在不同样本里能对齐
- 保留生物学差异:让不同条件(治疗 vs 对照)的真实差异不被一起抹掉
先想清楚批次的来源是哪一类:
| 类型 | 例子 | 整合策略 |
|---|---|---|
| 纯技术批次 | 同样品分两批跑、不同 chemistry | 强整合,应该完全对齐 |
| 个体差异 | 5 个健康人 PBMC | 强整合,假定细胞组成大同小异 |
| 条件差异 | 治疗 vs 对照、健康 vs 疾病 | 谨慎整合,对齐细胞类型但保留组间差异 |
| 物种差异 | 人 vs 鼠 | 跨物种工具(LIGER),普通方法不行 |
| 组织差异 | 肿瘤 vs 正常组织 | 看研究目标,如果想找新细胞类型反而不要整合 |
整合过度的代价:把治疗组特有的耗竭 T 细胞和对照组的正常 T 细胞强行拉到一起,下游差异分析就什么都看不到了。
批次效应从哪来
批次可以大致分两类:
- 技术性:不同测序批次、不同上机日期、不同试剂版本(chemistry v2 vs v3)、不同操作人员、不同处理流程。
- 生物学性:不同个体、不同组织、不同时间点、不同处理条件。这类本身也是想要研究的信号,所以"整合"时要特别小心别把它也去掉。
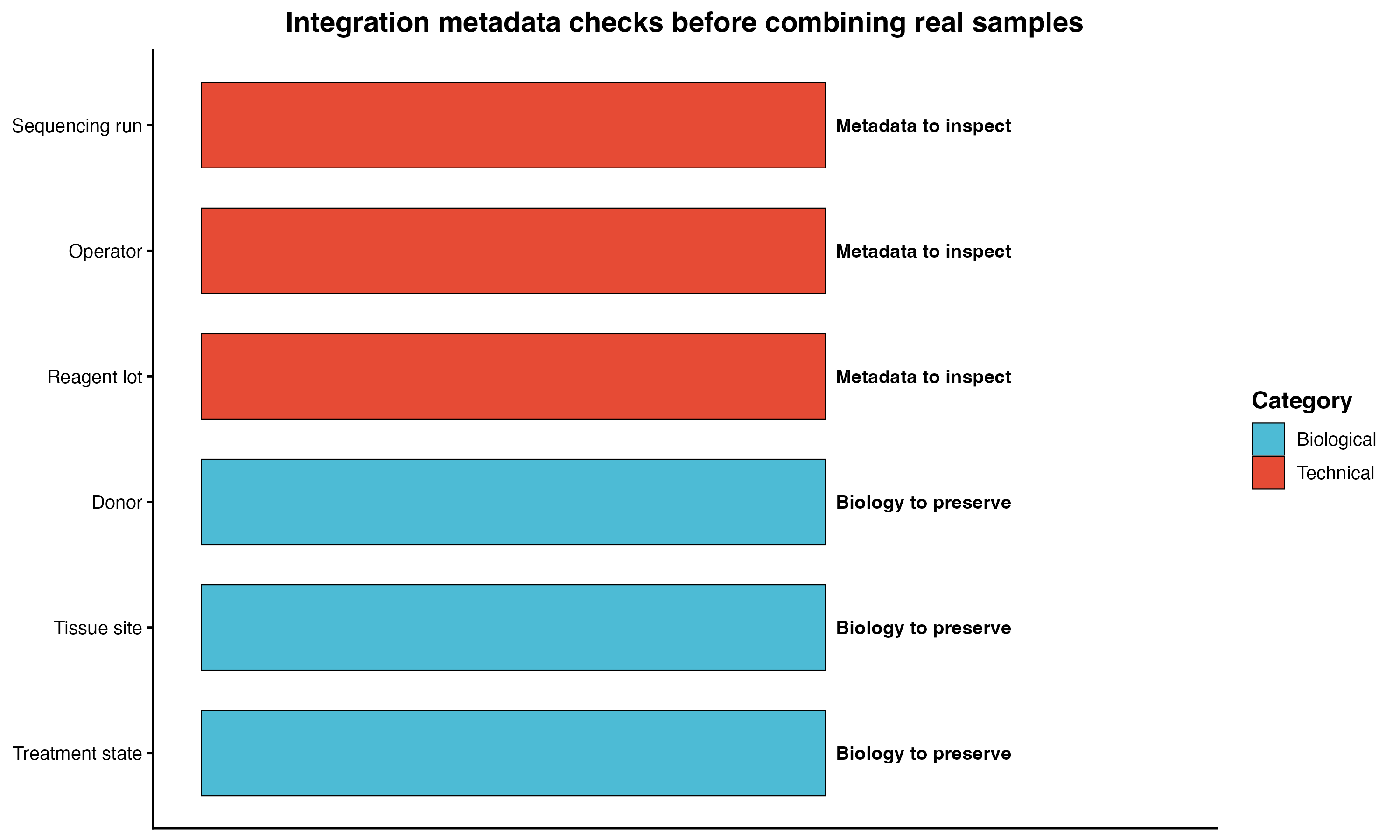
图 1:批次效应的来源。展示了技术批次和生物学批次的不同来源及其相对影响。
后果:如果不整合,不同批次的细胞会在 UMAP 上分开成两片,同一个细胞类型会被分到不同 cluster,差异分析里"条件 A vs 条件 B"就分不清是真实差异还是批次差异。
方法选择
| 方法 | 工具 | 速度 | 适合场景 |
|---|---|---|---|
| CCA / RPCA | Seurat | 中 | 样本间有充分共享的细胞类型 |
| Harmony | harmony(R/Py) | 快 | 大多数常规场景的首选 |
| scVI / scANVI | scvi-tools | 慢(GPU 快) | 样本量大、想做概率建模 |
| LIGER | liger | 中 | 跨物种、跨技术整合 |
| ComBat | sva | 快 | 仅批次校正,不做细胞层整合 |
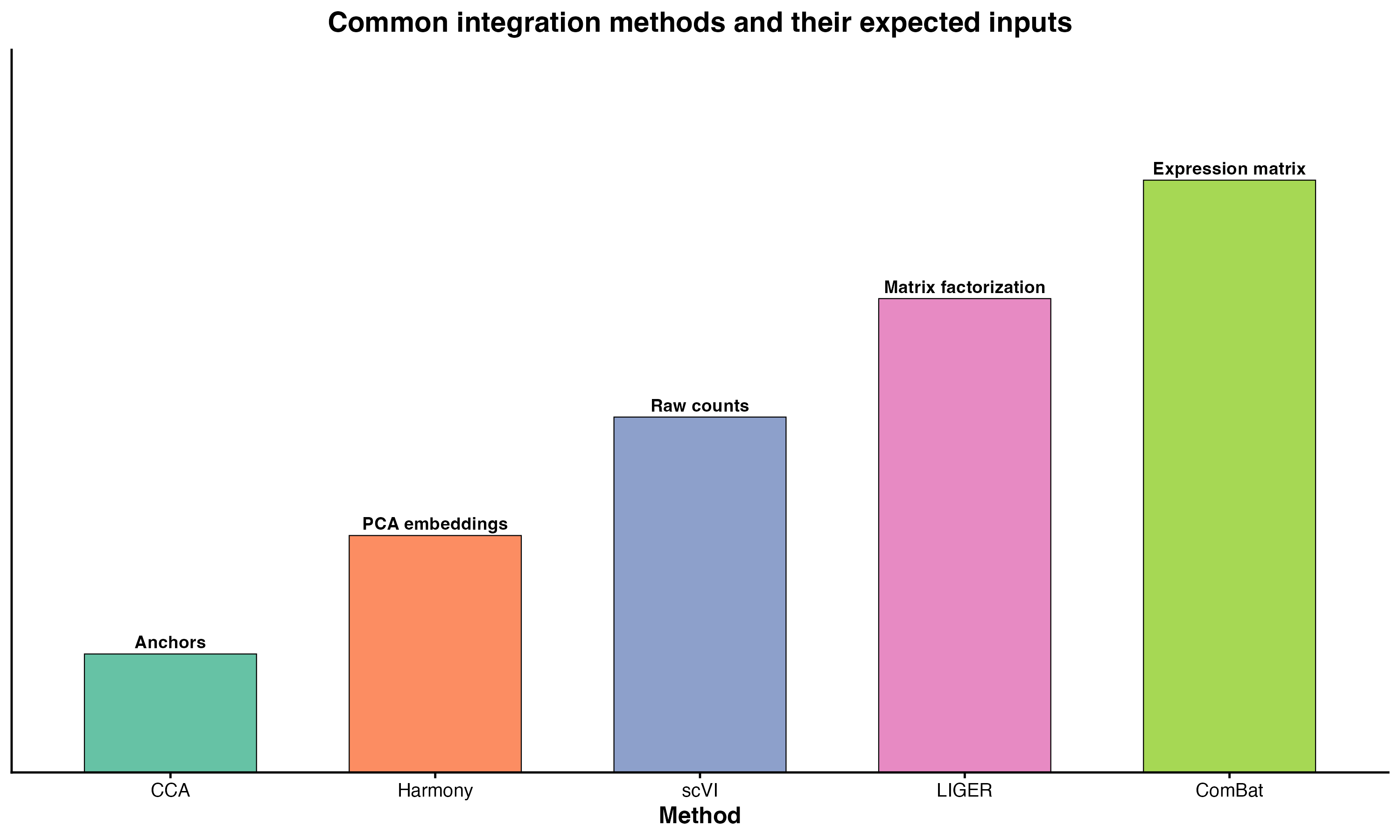
图 2:不同整合方法的对比。展示了 CCA、Harmony、scVI、LIGER 和 Combat 五种方法在速度、准确性和可扩展性三个维度的评分。
实际建议:两三个样本先试 Harmony。它快、对大多数场景够用、不需要 GPU。遇到 Harmony 整合过度或欠整合时再换 CCA/RPCA 或 scVI。
Seurat CCA 整合
Seurat 的整合分两步:各样本单独预处理 → 找 anchor → 在 integrated assay 上继续分析。
读入多个样本
library(Seurat)
sample1 <- Read10X("sample1/filtered_feature_bc_matrix/")
sample2 <- Read10X("sample2/filtered_feature_bc_matrix/")
sample3 <- Read10X("sample3/filtered_feature_bc_matrix/")
pbmc.list <- list(
sample1 = CreateSeuratObject(sample1, project = "sample1"),
sample2 = CreateSeuratObject(sample2, project = "sample2"),
sample3 = CreateSeuratObject(sample3, project = "sample3")
)
各自预处理
pbmc.list <- lapply(pbmc.list, function(x) {
x <- NormalizeData(x)
x <- FindVariableFeatures(x, selection.method = "vst", nfeatures = 2000)
x
})
找 anchor 并整合
features <- SelectIntegrationFeatures(object.list = pbmc.list)
anchors <- FindIntegrationAnchors(object.list = pbmc.list, anchor.features = features)
pbmc.combined <- IntegrateData(anchorset = anchors)
DefaultAssay(pbmc.combined) <- "integrated"
pbmc.combined <- ScaleData(pbmc.combined, verbose = FALSE)
pbmc.combined <- RunPCA(pbmc.combined, npcs = 30, verbose = FALSE)
pbmc.combined <- RunUMAP(pbmc.combined, reduction = "pca", dims = 1:30)
pbmc.combined <- FindNeighbors(pbmc.combined, reduction = "pca", dims = 1:30)
pbmc.combined <- FindClusters(pbmc.combined, resolution = 0.5)
DimPlot(pbmc.combined, reduction = "umap", group.by = "orig.ident")
DimPlot(pbmc.combined, reduction = "umap", label = TRUE)
两张图放一起看:第一张按样本上色,如果整合好了每个 cluster 里三个样本都均匀出现;第二张看 cluster 结构是不是符合预期。
图 3:PBMC 3k 真实细胞在 PCA 空间中的 barcode subset 分布。这里不伪造批次效应,只展示真实矩阵可计算出的坐标。
图 4:PBMC 3k 真实细胞在 UMAP 空间中的 barcode subset 分布。它用于演示观察子集混合情况的方法,不代表真实整合后的多样本结果。
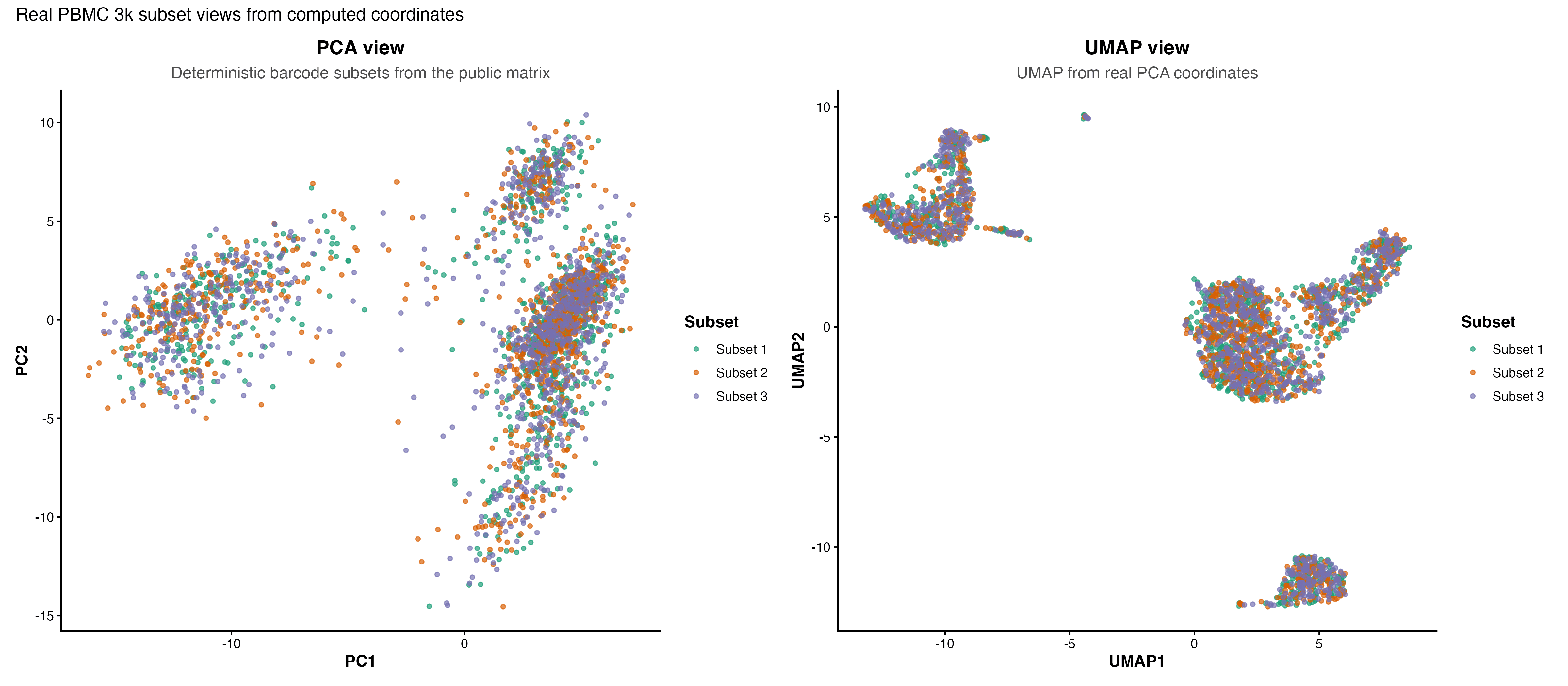
图 5:同一批真实 PBMC 3k 细胞的 PCA 与 UMAP 视图对比,用于说明整合诊断图应如何检查坐标、分组和细胞类型结构。
用 SCTransform �整合
SCTransform 版本:
pbmc.list <- lapply(pbmc.list, SCTransform)
features <- SelectIntegrationFeatures(object.list = pbmc.list, nfeatures = 3000)
pbmc.list <- PrepSCTIntegration(object.list = pbmc.list, anchor.features = features)
anchors <- FindIntegrationAnchors(
object.list = pbmc.list,
normalization.method = "SCT",
anchor.features = features
)
pbmc.sct <- IntegrateData(anchorset = anchors, normalization.method = "SCT")
pbmc.sct <- RunPCA(pbmc.sct, verbose = FALSE)
pbmc.sct <- RunUMAP(pbmc.sct, reduction = "pca", dims = 1:30)
Harmony 整合
Harmony 是目前最常用的快速整合方案。思路是直接在 PCA 之后对每个 PC 做批次校正,不新建 integrated assay,下游分析切到 reduction = "harmony" 就行。
library(harmony)
pbmc.merged <- merge(
pbmc.list[[1]], y = c(pbmc.list[[2]], pbmc.list[[3]]),
add.cell.ids = c("S1", "S2", "S3")
)
pbmc.merged <- NormalizeData(pbmc.merged) |>
FindVariableFeatures() |>
ScaleData() |>
RunPCA()
pbmc.harmony <- RunHarmony(pbmc.merged, group.by.vars = "orig.ident")
pbmc.harmony <- RunUMAP(pbmc.harmony, reduction = "harmony", dims = 1:30)
pbmc.harmony <- FindNeighbors(pbmc.harmony, reduction = "harmony", dims = 1:30)
pbmc.harmony <- FindClusters(pbmc.harmony, resolution = 0.5)
DimPlot(pbmc.harmony, reduction = "umap", group.by = "orig.ident")
DimPlot(pbmc.harmony, reduction = "umap", label = TRUE)
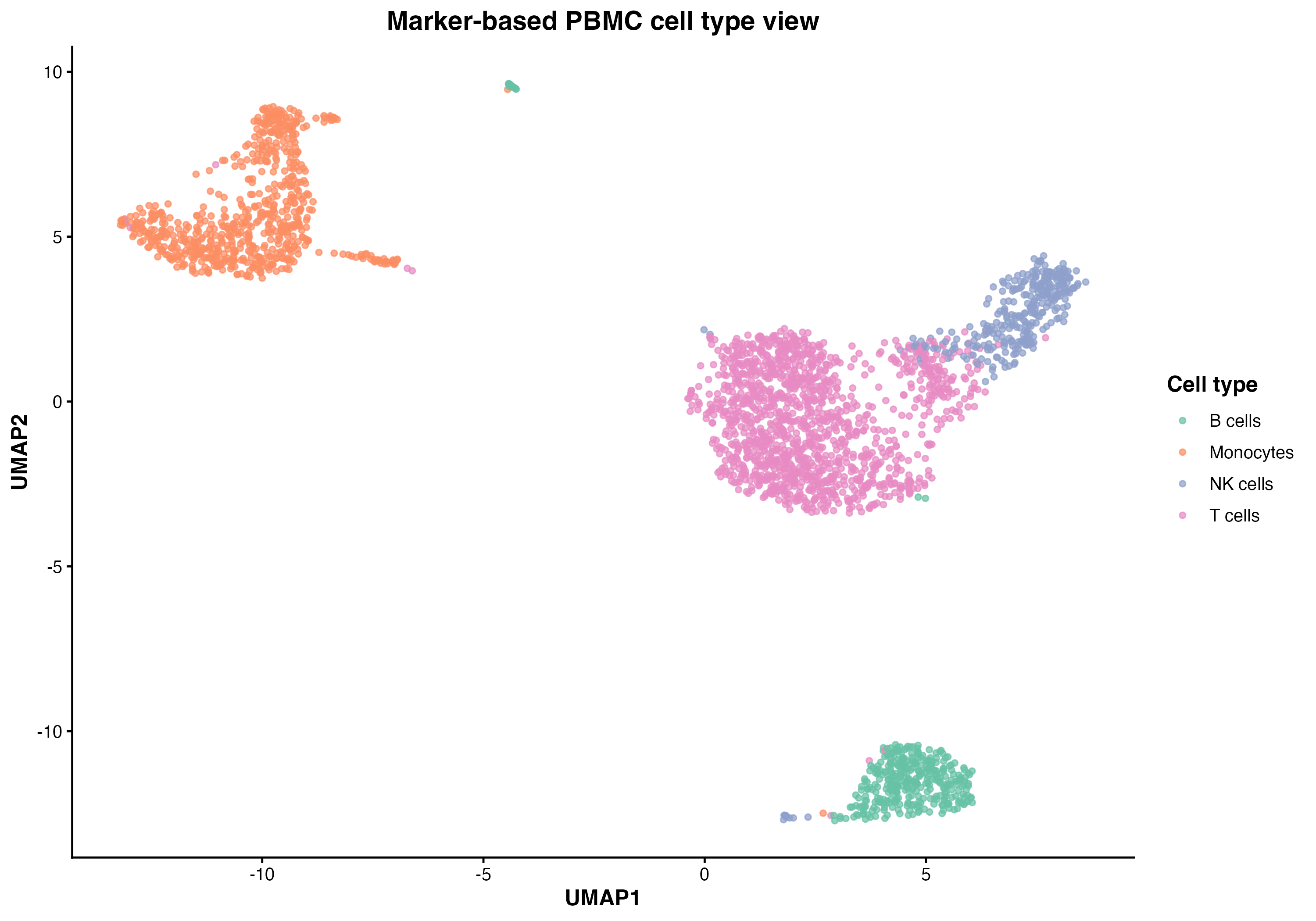
图 6:基于真实 marker 表达打分得到的 PBMC 细胞类型视图,展示 T 细胞、B 细胞、单核细胞和 NK 细胞在 UMAP 空间中的分布。
Python 端:scVI 或 Harmony
scVI
scVI 用变分自编码器做批次整合,样本多或想做概率建模时合适。在 GPU 上跑很快。
import scanpy as sc
import scvi
adatas = [
sc.read_10x_mtx("sample1/"),
sc.read_10x_mtx("sample2/"),
sc.read_10x_mtx("sample3/"),
]
for i, ad in enumerate(adatas, 1):
ad.obs["batch"] = f"sample{i}"
adata = adatas[0].concatenate(*adatas[1:])
sc.pp.filter_cells(adata, min_genes=200)
sc.pp.filter_genes(adata, min_cells=3)
sc.pp.normalize_total(adata, target_sum=1e4)
sc.pp.log1p(adata)
sc.pp.highly_variable_genes(adata, n_top_genes=2000, batch_key="batch")
scvi.model.SCVI.setup_anndata(adata, batch_key="batch")
model = scvi.model.SCVI(adata)
model.train()
adata.obsm["X_scVI"] = model.get_latent_representation()
sc.pp.neighbors(adata, use_rep="X_scVI")
sc.tl.umap(adata)
sc.tl.leiden(adata)
sc.pl.umap(adata, color=["batch", "leiden"])
Harmony (Python)
import scanpy as sc
import scanpy.external as sce
adata = adatas[0].concatenate(*adatas[1:])
sc.pp.normalize_total(adata)
sc.pp.log1p(adata)
sc.pp.highly_variable_genes(adata)
sc.pp.scale(adata)
sc.tl.pca(adata)
sce.pp.harmony_integrate(adata, "batch")
sc.pp.neighbors(adata, use_rep="X_pca_harmony")
sc.tl.umap(adata)
sc.tl.leiden(adata)
sc.pl.umap(adata, color=["batch", "leiden"])
怎么看整合有没有做好
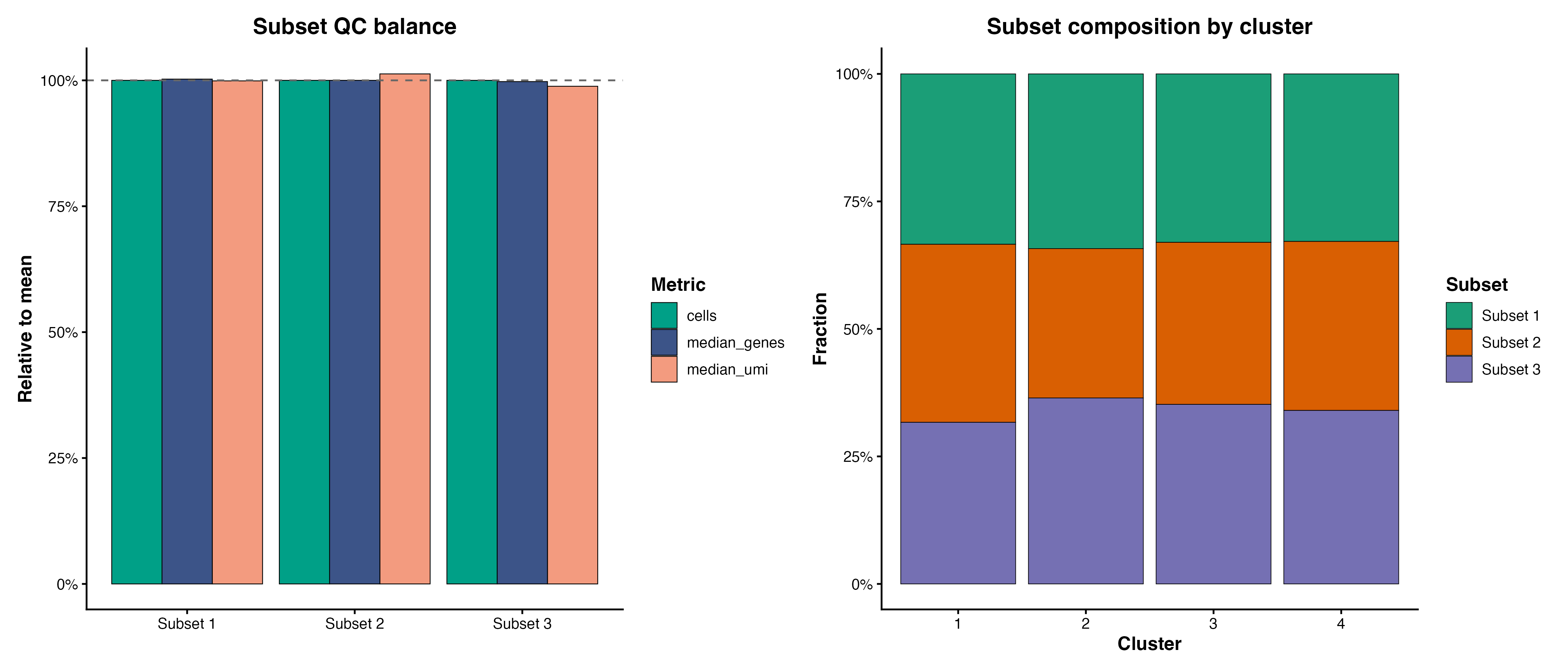
图 7:PBMC 3k barcode subset 的 QC 平衡和 cluster 组成。正式多样本项目可以把这里的 subset 替换成真实 sample 或 batch。
三件事要同时满足:批次混匀 + cluster 结构合理 + 生物学差异保留。
1. 批次混匀(visual)
p1 <- DimPlot(pbmc.merged, reduction = "umap", group.by = "orig.ident") +
ggtitle("整合前")
p2 <- DimPlot(pbmc.combined, reduction = "umap", group.by = "orig.ident") +
ggtitle("整合后")
p1 + p2
整合前三个样本会散成三块;整合后在同一 cluster 里三个样本应该均匀分布。
2. 定量评估:LISI
LISI(Local Inverse Simpson's Index)衡量"一个细胞周围邻居来自多少不同批次"。值接近样本数说明批次混匀,接近 1 说明邻居都来自同一批次(没整合好)。
library(lisi)
lisi_scores <- compute_lisi(
pbmc.combined@reductions$pca@cell.embeddings,
pbmc.combined@meta.data,
c("orig.ident")
)
summary(lisi_scores)
3. 生物学差异保留
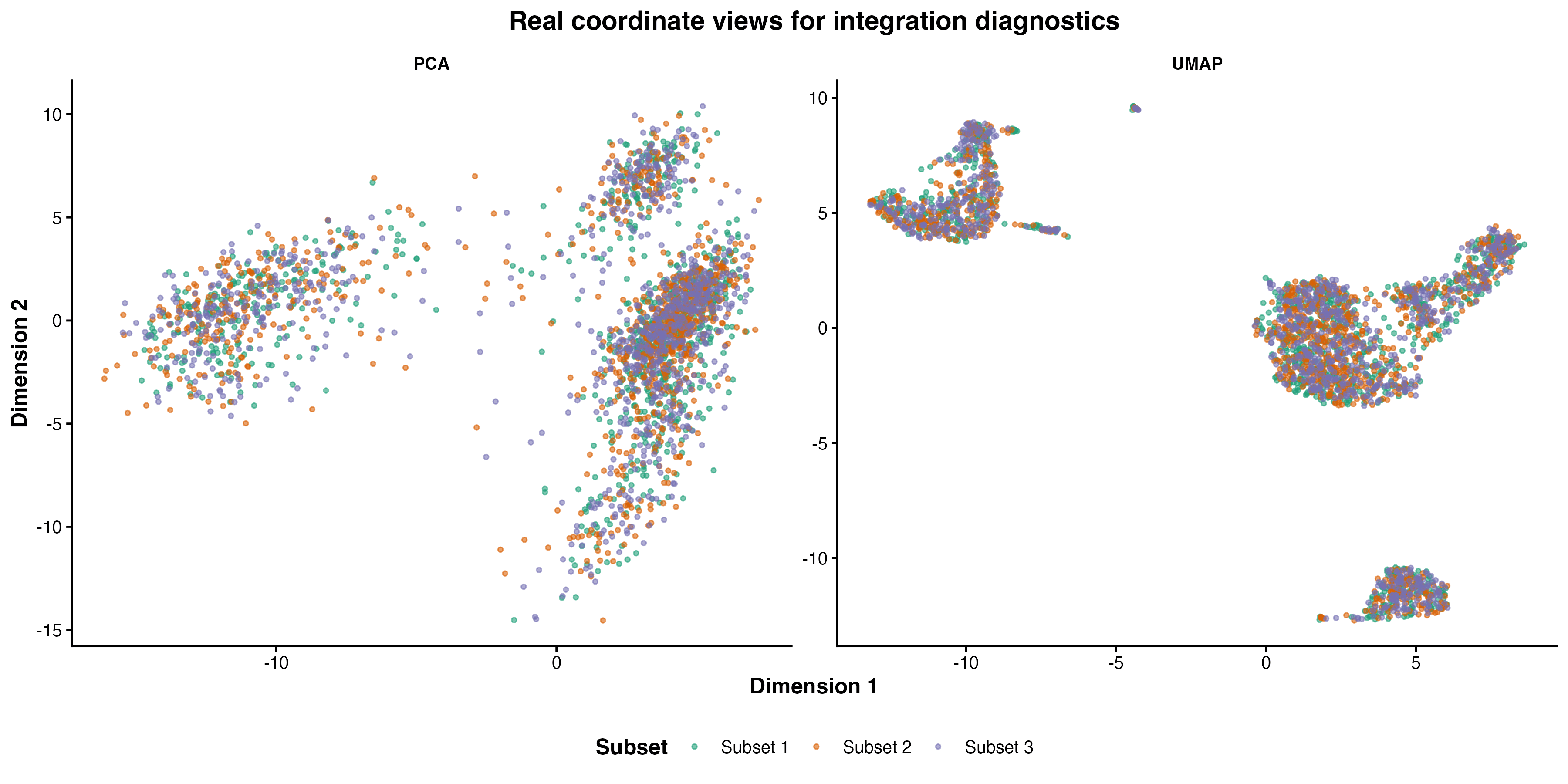
图 8:真实 PCA 与 UMAP 坐标下的 barcode subset 诊断视图。它展示图形检查方法,不声称比较多个整合算法的真实效果。
# marker 表达应该在对的 cluster 上
FeaturePlot(pbmc.combined, features = c("CD3D", "CD14", "MS4A1", "NKG7"))
# 如果有"治疗 vs 对照"这种生物学差异,splitsby 看一下是否被抹掉
DimPlot(pbmc.combined, split.by = "condition")
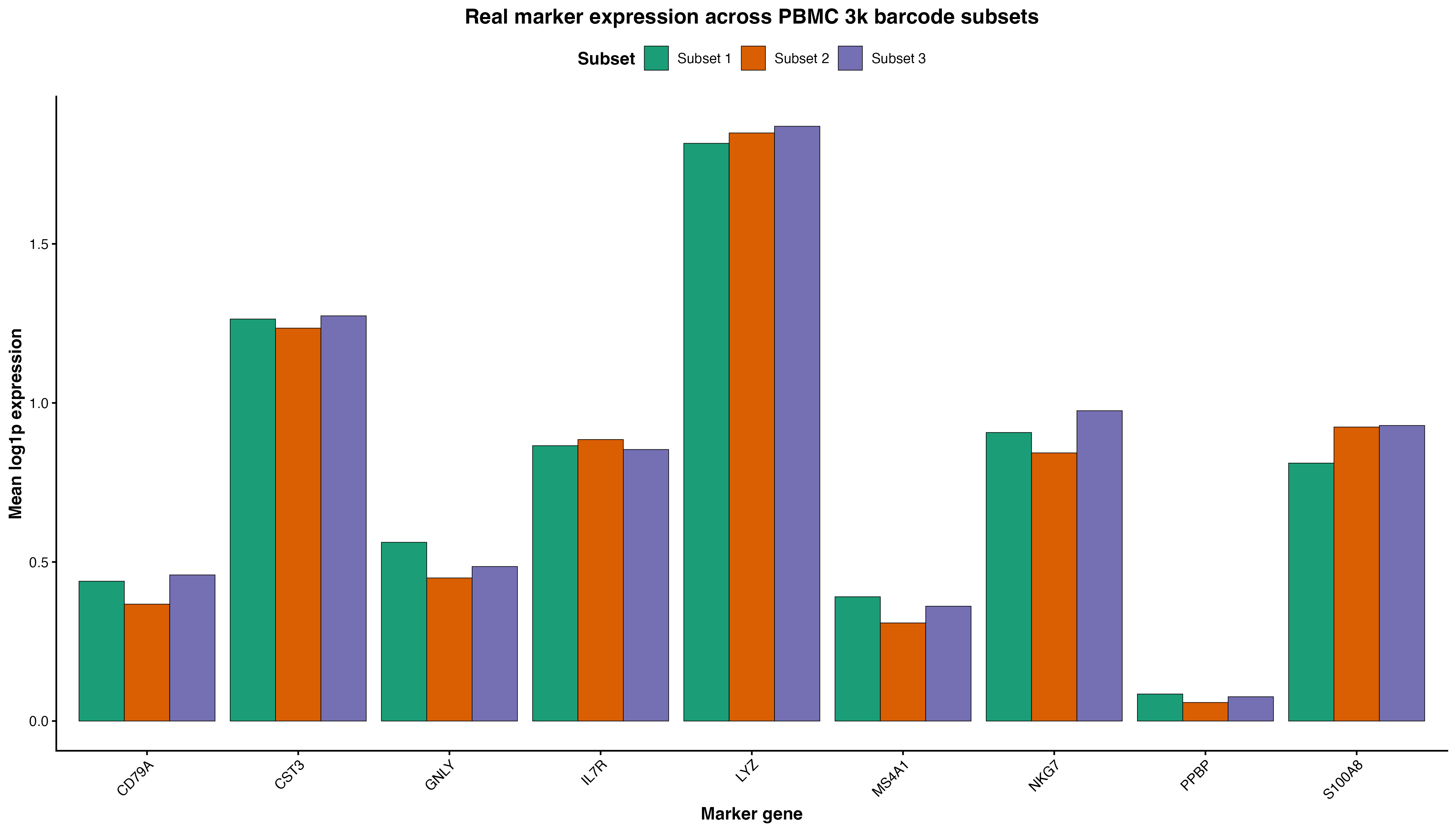
图 9:PBMC 3k marker 基因在真实 barcode subset 中的平均表达,用于检查分组之间 marker 信号是否被异常改变。
整合过度的典型表现:cluster 边界很乱、marker gene 不像该类细胞的 marker、同一条件 vs 不同条件的差异被抹平。遇到这种情况要回去调 k.anchor 或 k.weight,或者换 RPCA。
整合之后做差异分析
差异分析不能用 integrated assay(它经过线性重投影,表达值已经不是生物学意义上的表达了)。要切回 RNA(或 SCT):
DefaultAssay(pbmc.combined) <- "RNA"
Idents(pbmc.combined) <- "cell_type"
cd4_treated_vs_ctrl <- FindMarkers(
pbmc.combined,
ident.1 = "treated",
group.by = "condition",
subset.ident = "CD4 T",
test.use = "MAST"
)
head(cd4_treated_vs_ctrl)
常见做法:按 cell_type 分组 → 组内比较 treated vs control → 每个细胞类型各一份差异表。
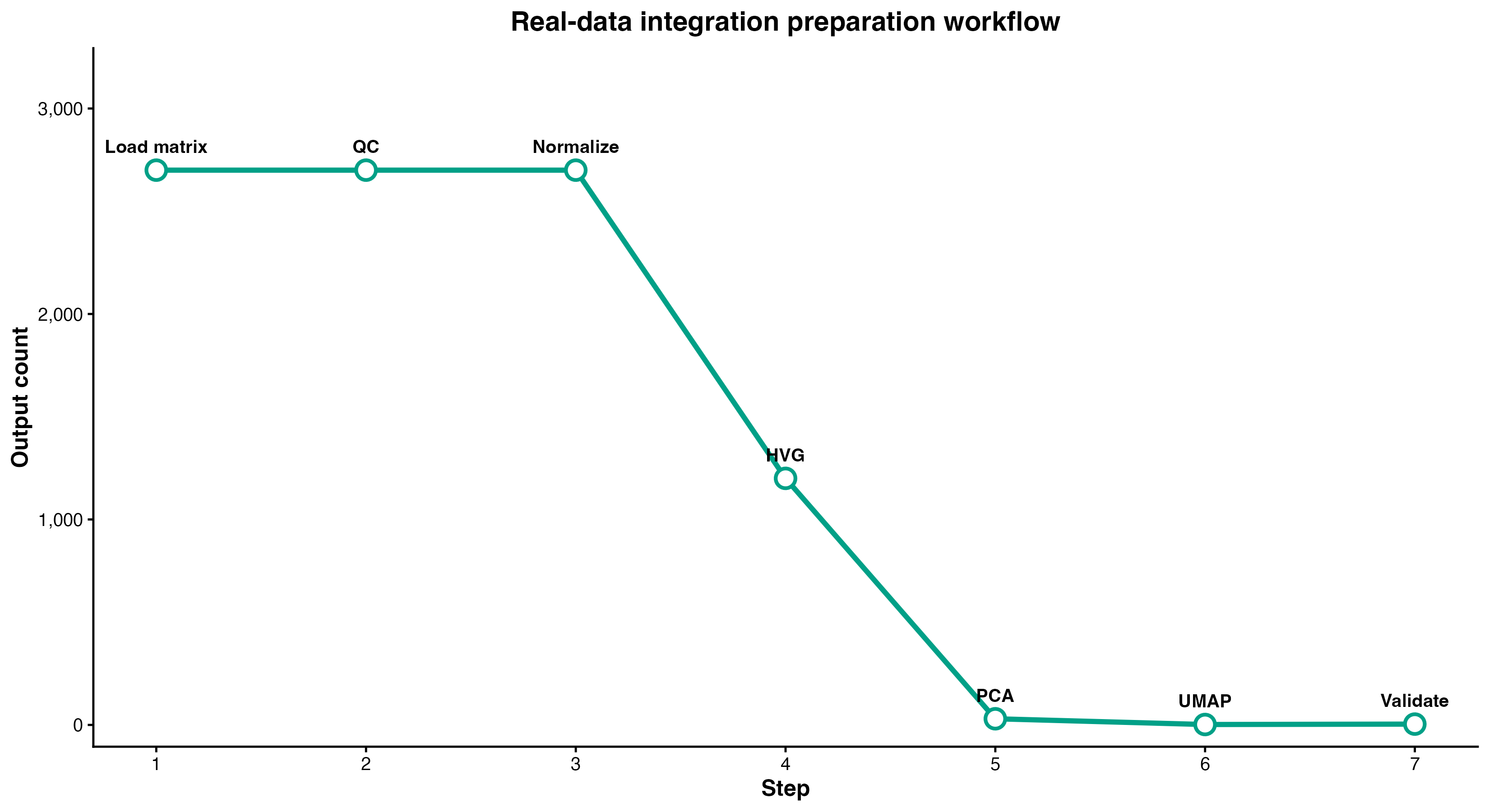
图 10:真实数据整合前的准备流程,展示从矩阵读取、QC、归一化、高变基因、PCA、UMAP 到验证的主要检查点。
常见坑
坑 1:先合并再做 QC
merge 之后才统一过滤,不同样本的质量差异会被掩盖。正确做法:每个样本各自做完 QC(线粒体阈值可以按样本调),再合并整合。否则一个低质量样本会拉低整体阈值。
坑 2:整合之后忘了切回 RNA assay 做差异分析
integrated 或 harmony 上的"表达值"是降维投影出来的伪值,跑 FindMarkers 出来的差异基因没有生物学意义。做差异分析必须 DefaultAssay(obj) <- "RNA"。
坑 3:把生物学差异当批次给整合掉
健康 vs 疾病、治疗 vs 对照的"组间差异"和"批次"是两回事。如果 group.by.vars = "sample" 把所有样本一律对齐,疾病组特有的细胞群就被抹平了。研究目标是组间比较时,整合的对象是"同一组内的不同个体",不是把不同组也合在一起。
坑 4:用了 SCTransform 又忘了 PrepSCTIntegration
SCTransform 路线整合需要先 PrepSCTIntegration 把各样本的 SCT model 对齐,否则 IntegrateData 会报错或给出错误结果。SCT 整合:先 SCT → PrepSCTIntegration → SelectIntegrationFeatures → FindIntegrationAnchors(normalization.method="SCT"),顺序不能错。
坑 5:Harmony 收敛就不管了,不查整合质量
Harmony 默认迭代到收敛即停止,但收敛 ≠ 整合好。一定要画 DimPlot(group.by="orig.ident"),看不同样本是否真的混匀了。混不匀就调 theta(默认 2,调大让整合更激进),或换 RPCA。
下载资源
下一步
接着深入:
- 05 轨迹推断与拟时序分析 — 多样本对齐之后,如果你的数据有连续过程(分化、应激响应),下一步是把"离散 cluster"变成"连续路径"
- 07 多模态数据分析 — 如果你的多样本里还包含蛋白 / ATAC,整合需要扩展到多模态
横向延伸:
- 03 质量控制、聚类与细胞类型注释 — 如果整合质量不好,常常是单样本 QC 没做好;回头检查这一步
- 整合方法基准评测论文(Luecken 2022) — 想深入了解各方法长短的读这篇
参考资源
离线资料下载
手册 HTML / PDF 已在后台预生成,点击后直接下载网站静态资源。