06 批次效应:ComBat / SVA / RUV
批次效应是 RNA-seq 里最容易让人上当的误差来源之一:同一天测完的样本可能更像;同一个技师做的样本可能更像;同一个 lane 出来的样本可能更像。真实的生物学差异如果被这种技术变量遮住,得到的"差异基因"就全是噪声。
处理批次有三条主流路线:
| 方法 | 思路 | 典型场景 |
|---|---|---|
| 把批次放进设计公式 | design = ~ batch + condition | 批次已知且与目标条件不完全混淆 |
| ComBat(sva 包) | 直接修改表达矩阵,把批次效应"减掉" | 批次已知;需要下游工具无法建模批次时 |
| SVA(Surrogate Variable Analysis) | 从表达矩阵里估计"看不到的"批次变量 | 批次未知,或还有其他技术变异 |
| RUVSeq | 用 control genes 估计不良变异 | 有 spike-in 或已知稳定基因时 |
优先级:能加进设计就加进设计(最干净)。设计不方便或者你不知道所有批次时再上 ComBat / SVA。
项目开始就该想的事:批次设计的预防
在数据已经测出来之后,再聪明的方法也只是补救。真正的批次问题应该在实验设计阶段就避免:
| 错误做法 | 后果 | 正确做法 |
|---|---|---|
| 第一周测对照、第二周测处理 | batch 和 condition 完全混淆,无法分离 | 每个 batch 都要含两组样本 |
| 同一个人只测一组 | 操作人成混杂因子 | 操作随机化或每人测同样比例 |
| 试剂买错版本(v2 vs v3) | chemistry 差异是技术 batch | 同一项目用同一批试剂 |
| 一个样本只跑一次测序 | 没法估技术重复噪声 | 至少几个样本做技术重复 |
| 上机日期是按组排的 | 上机批次就是组别 | randomize 上机顺序 |
最重要的一条规则:实验设计阶段 condition 和 batch 必须 balanced(平衡)—— 每个 batch 里每个 condition 都有样本。混淆了就是混淆了,统计上 unfixable。
经典教学例子:bladderbatch
bladderbatch 是 sva 作者提供的 microarray 数据,正因为批次和癌症状态重度混淆,成了批次校正教学的标配:
batch
cancer 1 2 3 4 5
Biopsy 0 0 0 5 4
Cancer 11 14 0 0 15
Normal 0 4 4 0 0
注意 batch 1、2、3 几乎全是某一类样本 —— 这就是"混淆设计"(confounded design)。混淆严重到一定程度,再聪明的方法也救不回来。但 bladderbatch 里留了足够的重叠,能看出不同校正方法的效果。
虽然是 microarray 数据,批次校正的原理和 RNA-seq 完全一样(数据经过 voom 或 VST 变换之后,两者本质上都是近似正态的连续数据矩阵),不妨碍教学。
真实示例:三种策略对比
配套脚本 bulk06_batch_sci.R 同时演示 bladderbatch 上的 ComBat / SVA / batch-in-design,以及 airway 上的"把 cell 当批次"。
Rscript scripts/bulk06_batch_sci.R
图 1:ComBat 前后的 PCA
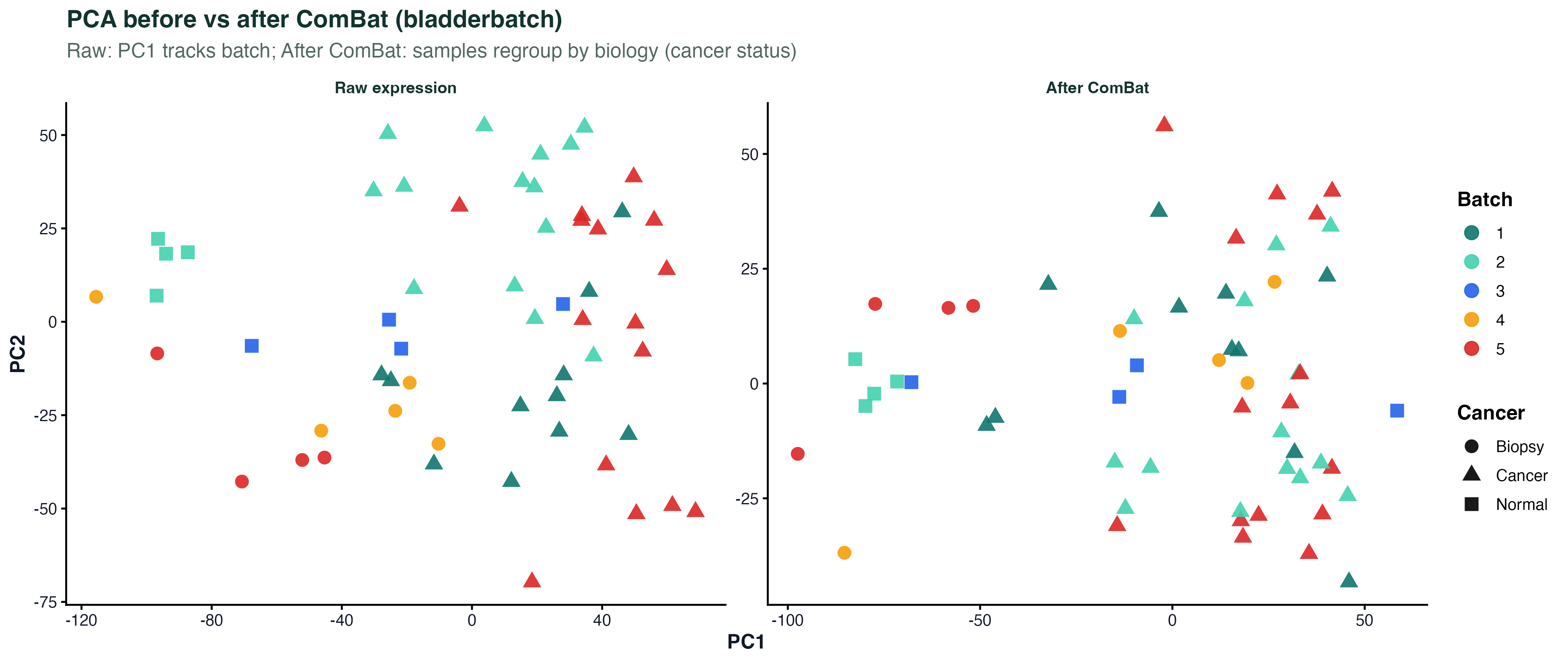
左边 raw:PC1 几乎被 batch 主宰 —— 同色的点(同批次)紧挨着,不同形状(癌症状态)散落在各处。右边 ComBat 之后:同形状的点(同癌症状态)开始靠拢,颜色(batch)反而变得弥散。
这张图把批次效应的本质直观化了:原始矩阵里批次变异 > 生物学变异。ComBat 做的就是把这个顺序掰回来。
library(sva)
mod <- model.matrix(~ as.factor(cancer), data = pheno)
combat_edata <- ComBat(dat = edata, batch = batch, mod = NULL)
mod 这里传 NULL 表示"不保护任何生物学变量",保守用法。如果确实担心 ComBat 把生物信号一起抹掉,可以传入 mod = model.matrix(~ cancer, ...),告诉 ComBat "这列是要保留的"。
图 2:样本距离热图
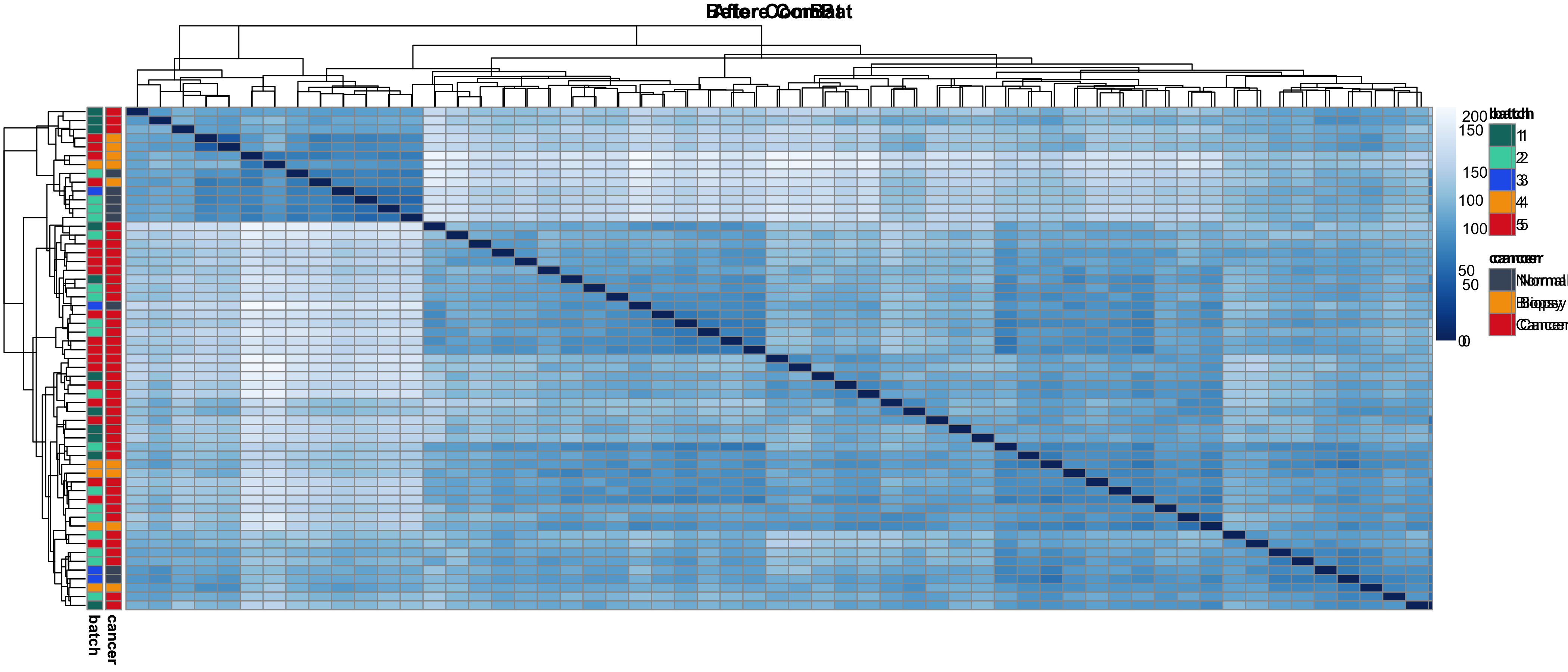
跟 PCA 互补的角度:样本两两之间的欧式距离。校正前样本按 batch 聚在一起;校正后按 cancer 状态聚。写 Methods 段时把 PCA + 距离热图一起放,说服力会更强。
图 3:SVA 估计出的隐变量
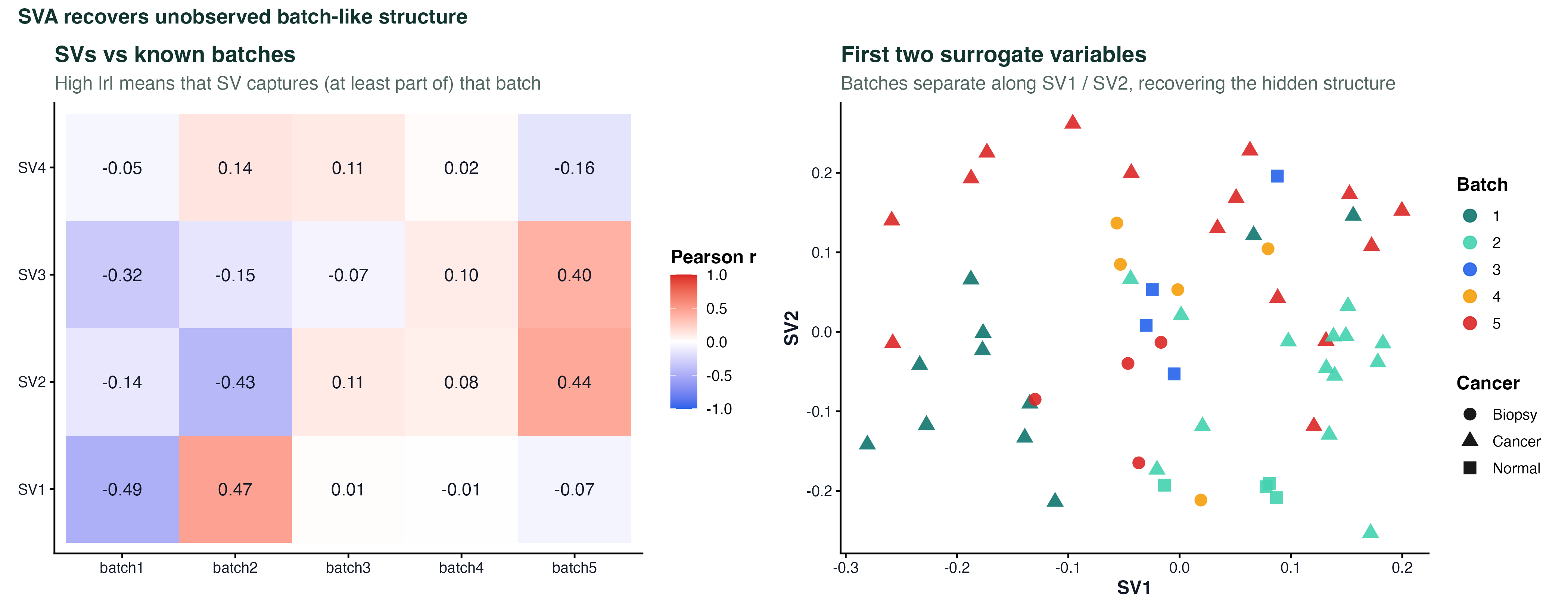
SVA 不依赖 batch 标签 —— 它直接从表达矩阵里估计"和生物学变量无关的系统性变异",结果叫 surrogate variable (SV)。
左图是 SV 和已知 batch 的相关性矩阵。每个 SV 都和 某一个 batch 相关性很强 —— 意味着 SVA 成功把隐藏的批次结构找出来了。右图把 SV1 vs SV2 画成散点,点按真实批次着色:同批次的样本在 SVA 空间里也聚到一起。
mod0 <- model.matrix(~ 1, data = pheno)
n_sv <- num.sv(edata, mod, method = "leek")
svobj <- sva(edata, mod, mod0, n.sv = n_sv)
# 把 svobj$sv 当成新列加进 limma / DESeq2 的设计矩阵
SVA 什么时候用:完全不知道 batch、或者知道 batch 但怀疑还有别的隐变量(试剂批次、操作人、机器 drift)时。
图 4:三种策略的 DE 基因数
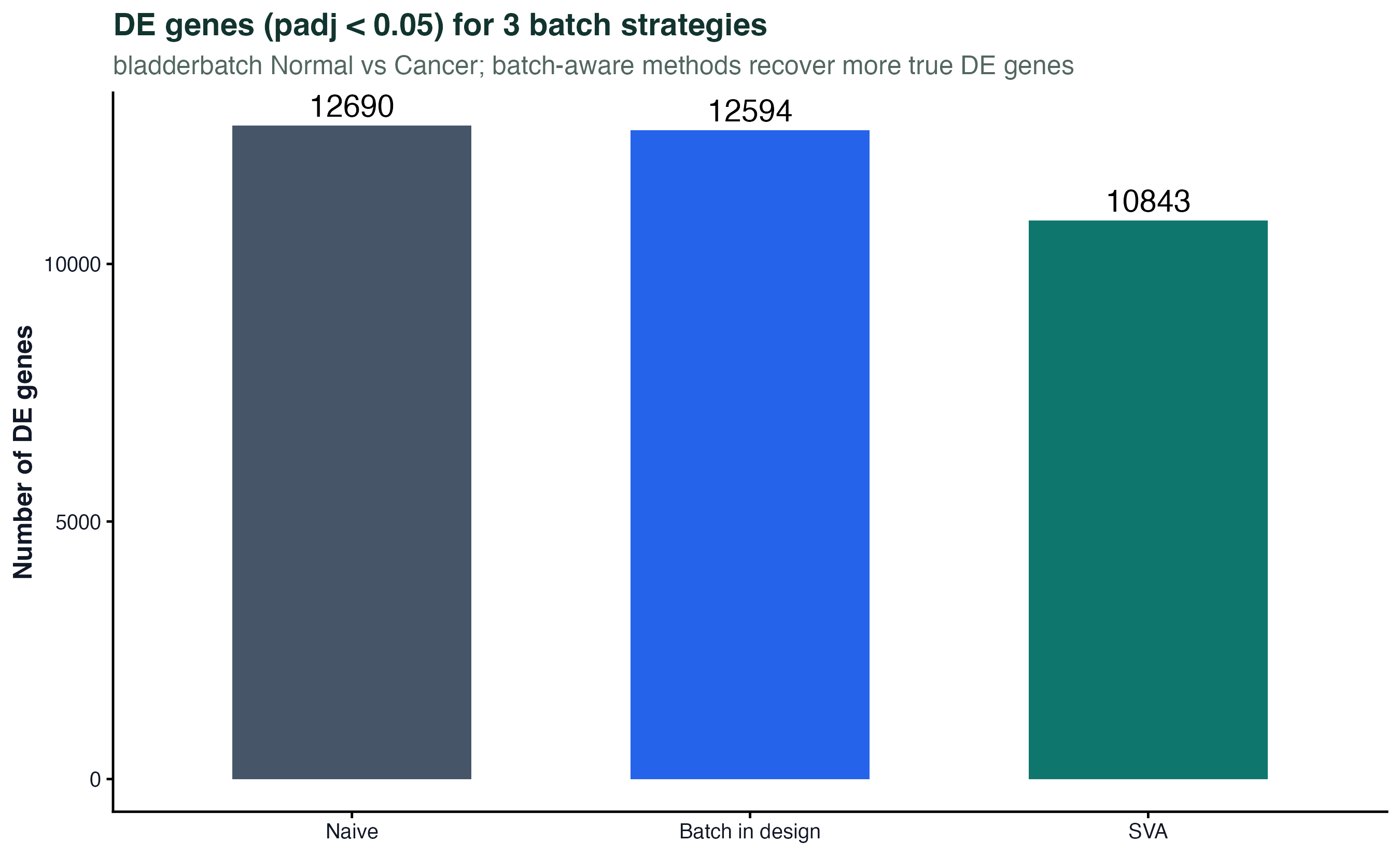
对 bladderbatch 里 Normal vs Cancer 的比较,三种策略(naive、把 batch 加进设计、用 SVA)分别跑 limma,统计 padj < 0.05 的差异基因数:
- Naive(忽略批次):显著基因数被批次噪声稀释,本来该显著的信号被掩盖
- Batch 放进 design:效力显著提高
- SVA:进一步挖出额外的隐变量,效力最高
这张图最直接地说明批次校正不是在"变出"基因,而是在"让真正的生物学信号从噪声里浮出来"。
图 5:三种策略的 DE 基因重叠
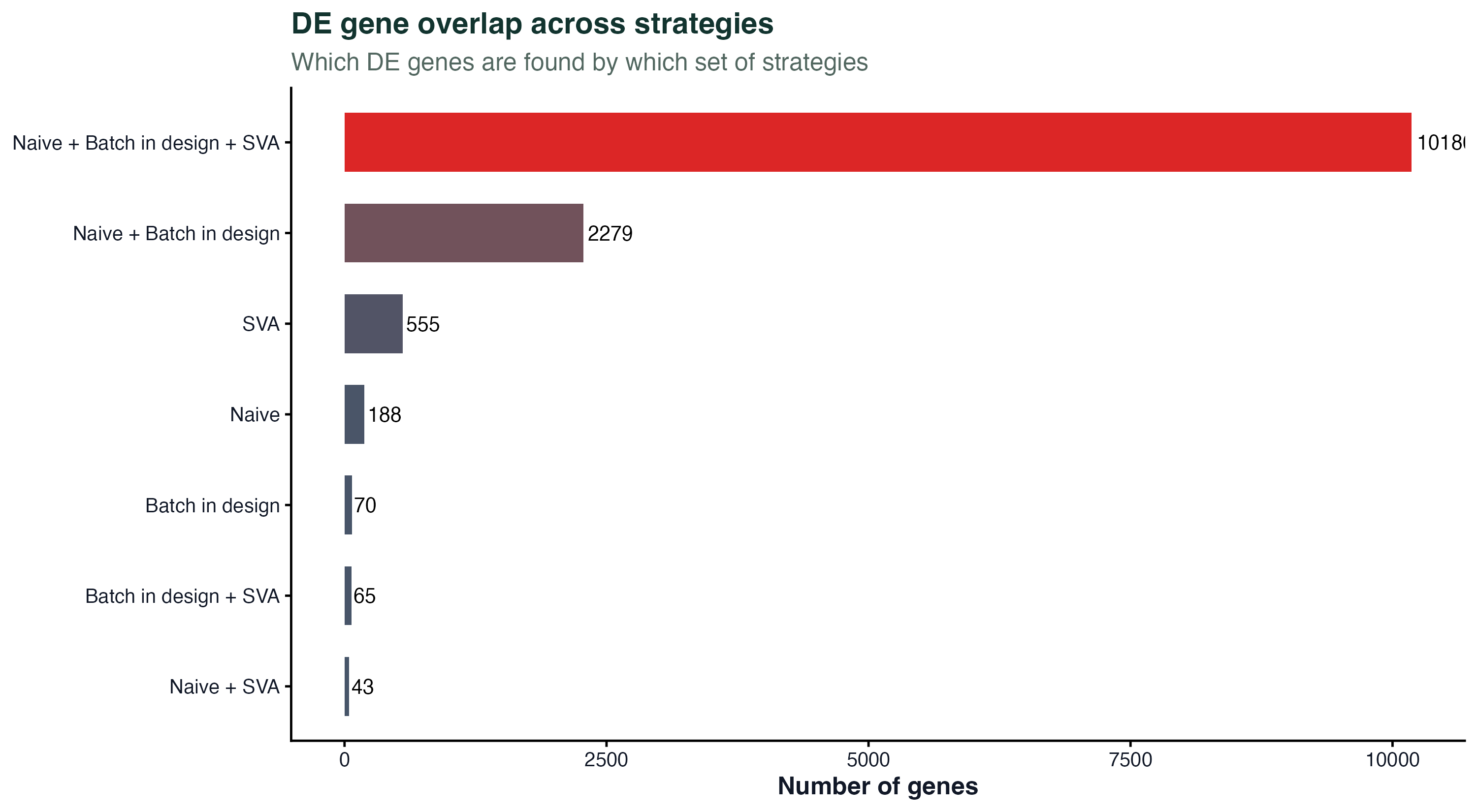
DE 基因列表在三种策略之间的重叠。最重要的是检查:naive 独有的那一部分基因,会不会其实是批次带来的假阳性? 同时看三种策略都发现的基因是信号最稳的那批,适合重点关注。
图 6:airway 里把 cell 当批次
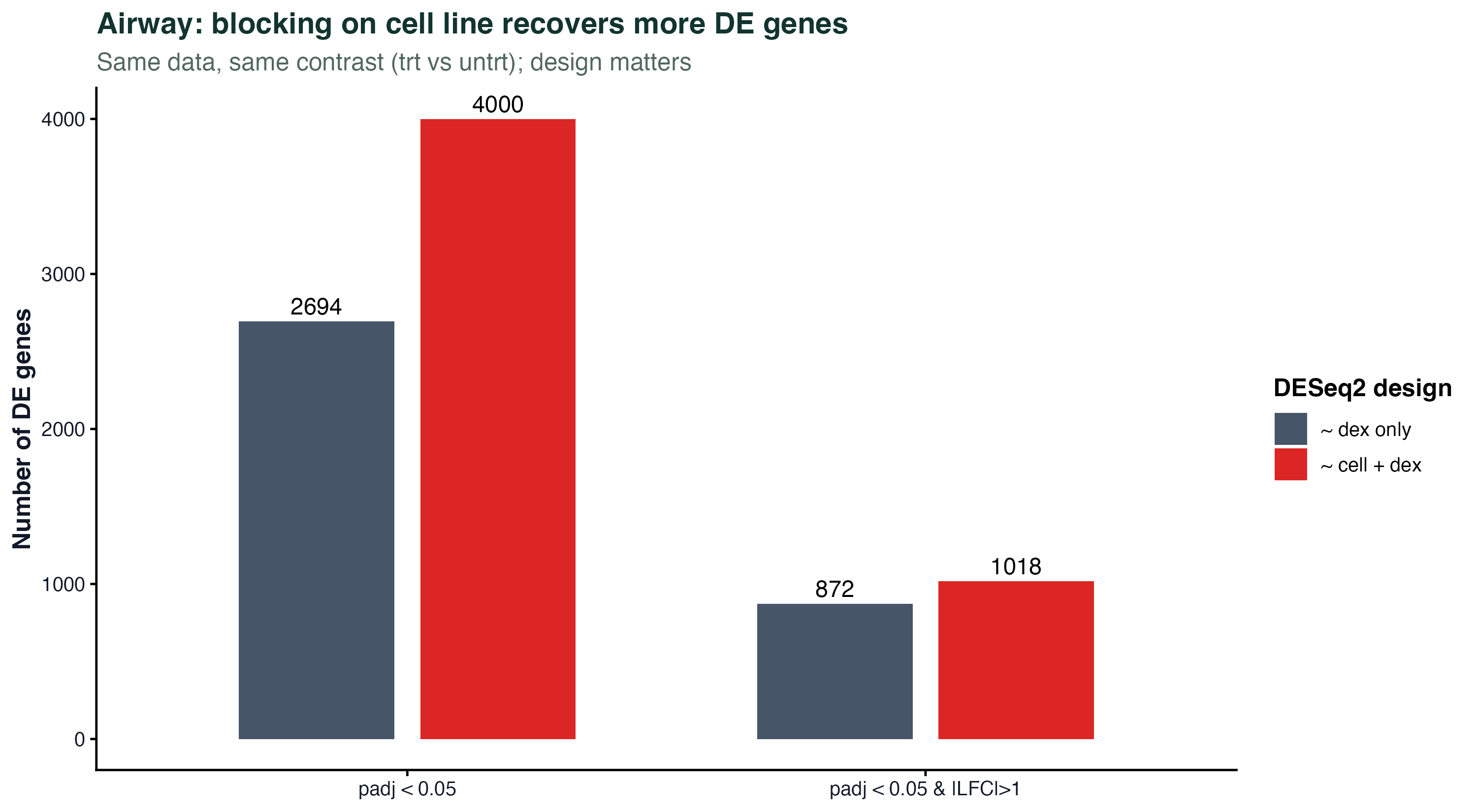
airway 有 4 个细胞系,每个细胞系都有 trt 和 untrt 两个样本。细胞系之间的基线表达差异是"技术批次"的一种 —— 只不过它是已知的。
design = ~ dex(忽略 cell):把细胞系间的变异算进残差,误差估得偏大,DE 基因变少design = ~ cell + dex(把 cell 放进去):细胞系间的基线被模型吃掉,检验 dex 效应的功效更高
在 padj < 0.05 & |LFC|>1 这个实用阈值下,把 cell 加进 design 可以把 DE 基因数从几百个提到数千个。这说明 DESeq2 里"batch 放不放进 design"不是个形式问题,是真的能改变结果的。
如何选择策略
实用建议:
| 情形 | 推荐做法 |
|---|---|
| batch 已知、未与条件混淆 | ~ batch + condition |
| batch 已知、部分混淆 | 同上 + 仔细检查 PCA / distance |
| batch 已知、完全混淆 | 设计本身有问题,尽量重测,其次 SVA |
| batch 未知 | SVA,估出 SV 之后加进 design |
| 有 spike-in 或 housekeeping | RUVg 或 RUVs |
| 下游工具不接受 design(老版 GSVA 之类) | ComBat 直接校正矩阵 |
绝对不要做的事:在已经很干净的数据上"保险起见跑一遍 ComBat"。ComBat 会让样本间的真实生物学变异也变平,显著增加 type II error。
和单细胞整合的关系
单细胞实践 04 多样本整合 里的 Harmony / Seurat v5 integration 思路和这里一致:在某个隐空间里把批次的那一部分方向抹掉,保留生物学方向。只不过单细胞是在 PCA 空间上做,bulk 是在原始 log 表达矩阵上做。
常见坑
坑 1:批次和条件完全混淆还硬上 ComBat
如果你的 batch 1 全是 control、batch 2 全是 treatment,ComBat / SVA / 任何方法都救不回来。模型分不清"组间差异"和"批次差异"。这种情况只能重新设计实验。用 table(batch, condition) 先看一眼分布,如果某行某列全是 0,停止分析。
坑 2:ComBat 不传 mod 参数
ComBat(dat, batch) 不传 mod 时是"保守模式",假定所有变异都是批次。但如果生物学差异恰好和批次有点相关(比如 batch 1 的样本恰巧大多是处理组),ComBat 会把生物学信号也当批次抹掉。正确做法 ComBat(dat, batch, mod = model.matrix(~ condition)),告诉它哪一列要保留。
坑 3:SVA 估出的 SV 数量过多
num.sv(method = "leek") 在小样本数据里有时给出 10+ 个 SV,全加进 design 会消耗自由度、降低统计力。SV 数量经验上 ≤ samples/3,超过这个先回头检查数据本身有没有别的问题。
坑 4:用 ComBat 之后还把 batch 加进 design
ComBat 已经把 batch ��信号从矩阵里去掉了,DESeq2 / limma 的 design 不应再含 batch(含了会双重校正)。ComBat → design = ~ condition;不用 ComBat → design = ~ batch + condition。两选一,不能同时用。
坑 5:以为 batch 校正后样本之间所有差异都是生物学
校正完看 PCA 漂亮了 — 这只是说明"batch 这个变量被消了",不代表"剩下的差异全是生物学"。可能还有别的混杂因子(性别、年龄、采样时间)。校正后还是要逐个 metadata 列做 PCA 染色,确认没有别的隐藏批次。
下载资源
下一步
接着深入:
- 07 多工具对比 — DESeq2 / edgeR / limma 在批次校正后的结果会有差别吗?
- 08 可复现分析报告 — Methods 段怎么写清楚 batch 处理方式
横向延伸:
- 单细胞 04 多样本整合 — 单细胞里 Harmony / Seurat integration 是同一思路在 PCA 空间的版本
- Leek 2010 batch effects 综述 — 批次效应统计学背景,是经典阅读
参考资源
离线资料下载
手册 HTML / PDF 已在后台预生成,点击后直接下载网站静态资源。