module03
速答
Q: ORA 和 GSEA 有什么区别? A: ORA 需要先筛差异基因(有阈值),再看基因集是否富集;GSEA 不筛阈值,用全部基因排序看基因集在排序里的分布。GSEA 更灵敏,能发现 ORA 漏掉的温和变化。
Q: 基因 ID 转换 SYMBOL / ENSEMBL / ENTREZID 哪个用?
A: clusterProfiler 的 enrichGO / gseKEGG 需要 ENTREZID;画图展示用 SYMBOL。用 clusterProfiler::bitr() 或 AnnotationDbi::mapIds() 转换。
Q: GO 的 BP / MF / CC 选哪个? A: 生物学过程(BP)最常报告,分子功能(MF)和细胞组分(CC)作为补充。三者都跑,重点解读 BP。
Q: GSEA 的 NES 和 pvalue 怎么解读? A: NES(Normalized Enrichment Score)> 1 正向富集,< -1 负向富集;|NES| 越大越显著。pvalue < 0.05 或 padj < 0.25 视为显著。
03 功能富集:GO / KEGG / GSEA
差异基因本身只是中间产物。真正有用的是"这些差异基因对应哪些通路、哪些功能"。功能富集分析用的就是已经注释好的基因集合,看差异基因列表和这些集合有没有非随机的重叠。
本章用 02 DESeq2 差异表达 里 airway 的差异结果,走一遍最常用的三种富集:
- GO(Gene Ontology):基因的生物过程 / 分子功能 / 细胞组�分
- KEGG:手工整理的代谢和信号通路
- GSEA(Gene Set Enrichment Analysis):不依赖阈值,直接看排序里的分布
ORA vs GSEA 两种思路
这两种方法是互补的,值得花一段说清楚它们的区别。
| 维度 | 过表达分析(ORA) | 基因集富集(GSEA) |
|---|---|---|
| 输入 | 一张"显著差异基因"名单 | 所有基因的排序(按某个 statistic) |
| 需要阈值 | 需要(padj < 0.05、|LFC| > 1 之类) | 不需要 |
| 擅长回答 | 强效应基因扎堆在哪些通路 | 整条通路整体是否往一个方向偏 |
| 典型工具 | enrichGO、enrichKEGG | gseGO、fgsea |
| 弱点 | 阈值非常敏感 | 对排序 statistic 选择敏感 |
工程上的建议:两种都跑,交叉印证。ORA 给出"非常确定的、强效应的通路";GSEA 能抓到那些"每个基因效应不大,但整条通路一致下降"的情况(比如线粒体电子传递链)。
举一个具体例子说明为什么两种都要跑:
某次实验做药物处理 vs 对照。用 padj < 0.05 & |LFC| > 1 阈值得到 50 个 DE 基因。
- ORA 结果:top 通路 "免疫激活"、"细胞凋亡" — 来源是 50 个 DE 基因里恰好有 8 个属于这两条通路
- GSEA 结果:top 通路 "线粒体电子传递链 (OXPHOS)" — 这条通路上 100 多个基因每个 LFC 只有 0.3-0.5,没一个过阈值,但整条通路一致下调
这种"分散的、每个基因小而全通路一致"的信号,ORA 完全看不见。OXPHOS 在很多药物处理项目里的下调正是这种类型。漏掉 GSEA 等于漏掉这一类机制。
基因 ID 的坑
clusterProfiler 的 enrichKEGG、enrichGO 都要求 Entrez ID 或能通过 OrgDb 映射到 Entrez。airway 的 counts 矩阵用的是 Ensembl gene ID(ENSG00000000003 这种),需要先做一步转换:
library(org.Hs.eg.db)
library(AnnotationDbi)
id_map <- AnnotationDbi::select(
org.Hs.eg.db,
keys = rownames(dds),
keytype = "ENSEMBL",
columns = c("ENTREZID", "SYMBOL")
)
一个 Ensembl ID 可能对应多个 Entrez ID(历史重命名、重复注释),也可能对应 NA。做富集前要 distinct() 和 filter(!is.na()) 清理一遍。
真实示例:airway 上的 GO + KEGG + GSEA
配套脚本 bulk03_enrichment_sci.R 接着上一章 DESeq2 的结果走:把 DE 结果的 Ensembl 映射成 Entrez → 对显著 DE 基因做 GO BP/MF/CC 和 KEGG 的 ORA → 对全部基因按 sign(LFC) * -log10(p) 排序做 GSEA。
Rscript scripts/bulk03_enrichment_sci.R
图 1:GO BP 富集 dotplot
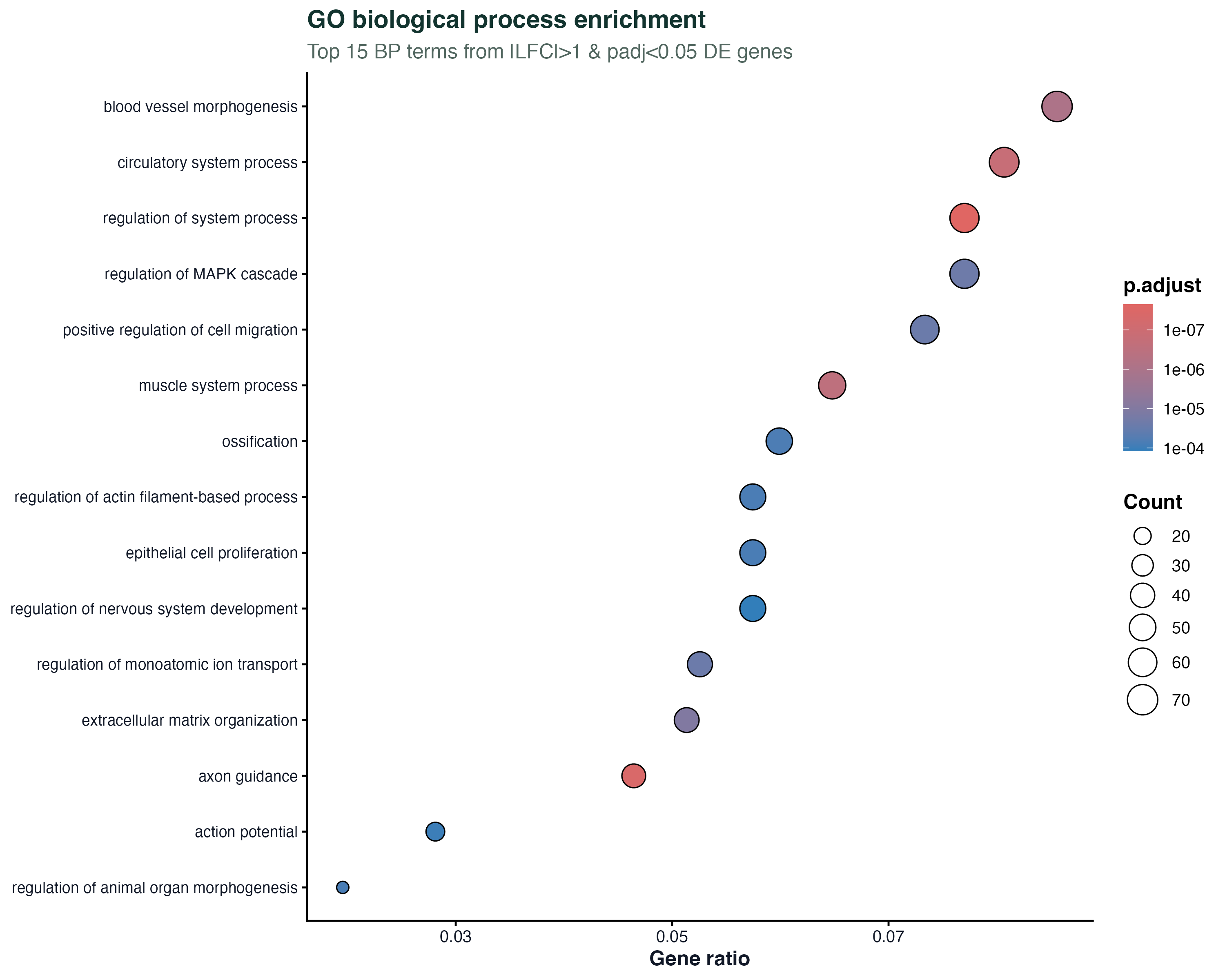
显著 DE 基因��在 GO biological process 里的 top 15 个 term。横轴 Gene ratio 是"term 里命中的 DE 基因数 / 输入的 DE 基因总数",点大小也代表命中数量,颜色是 p.adjust。
airway 是地塞米松处理的气道上皮,跑出来能看到典型的糖皮质激素响应:类固醇激素响应、细胞增殖调控、上皮细胞分化之类。这种"药理作用 → 预期通路"的对应就是富集分析最直观的验证。
图 2:GO 三大类(BP/MF/CC)的 top 条目
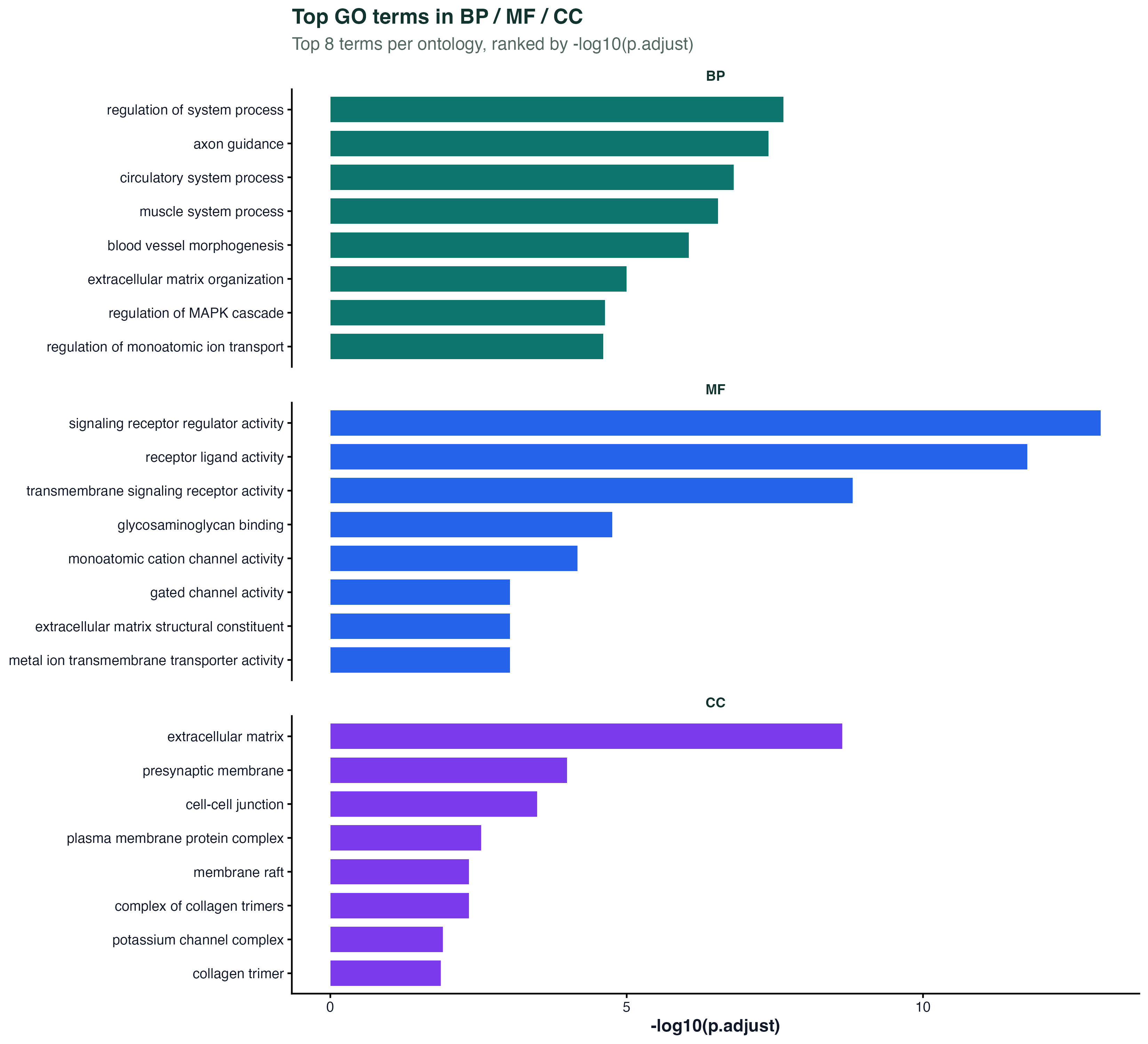
GO 分三个子本体:BP(生物过程)、MF(分子功能)、CC(细胞组分)。三个一起看能给出更全的解读:BP 告诉你"在做什么",MF 告诉你"通过什么分子机制",CC 告诉你"主要发生在哪里"。
图里每个子本体取 top 8,颜色区分。写 Methods 段的时候这张图能同时列出三类核心 term,比单独看 BP 更完整。
图 3:GO term 相似度网络
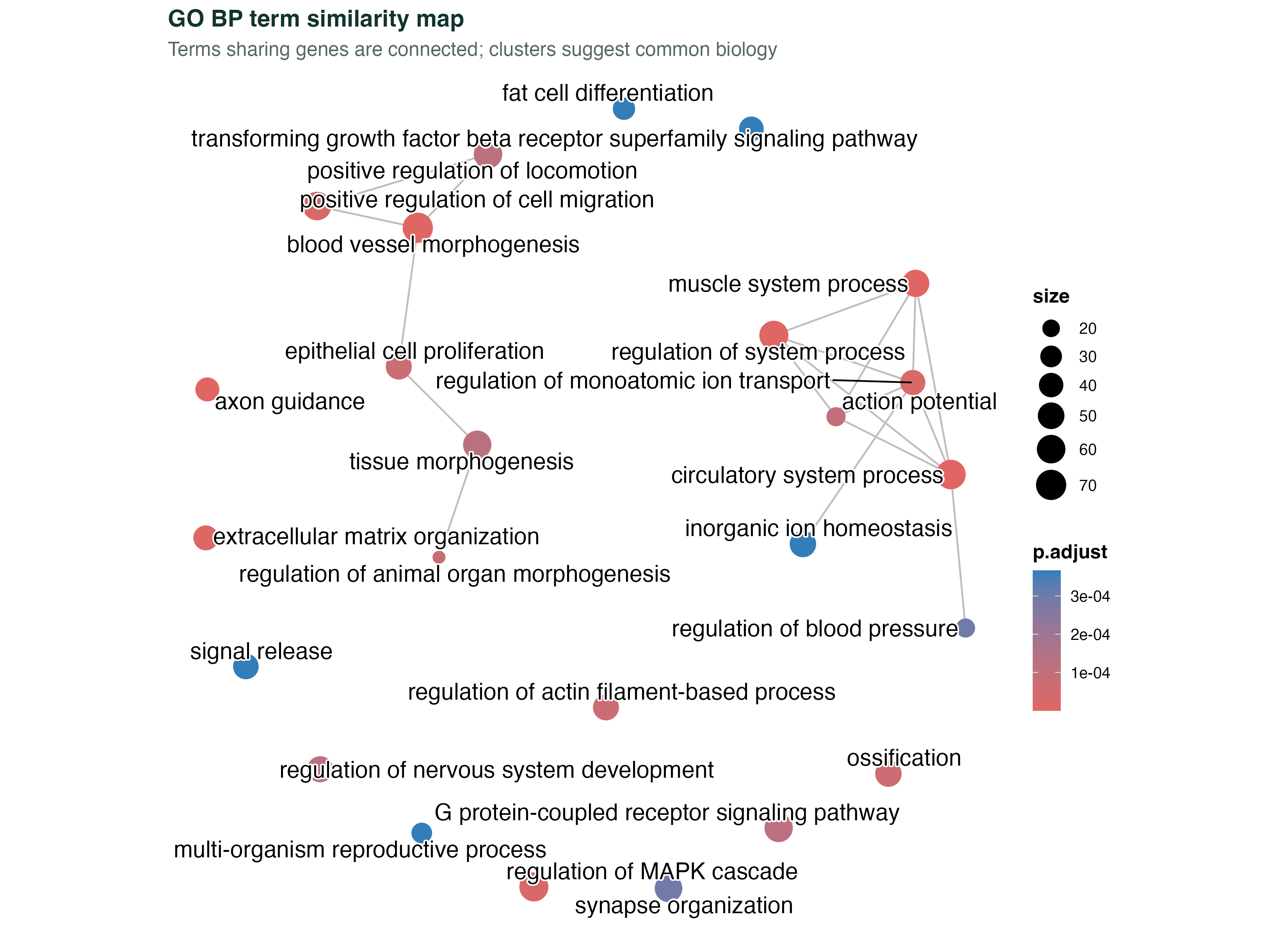
很多 GO term 之间共享基因(��比如 "regulation of cell proliferation" 和 "positive regulation of cell proliferation")。emapplot 把 term 之间的基因重叠画成网络,聚在一起的 term 通常讲的是同一组生物学。
这张图的价值是去冗余:直接看 top 15 dotplot 容易发现很多 term 其实在说同一件事;emapplot 能帮你把它们归成几个有意义的簇。
图 4:KEGG pathway 富集
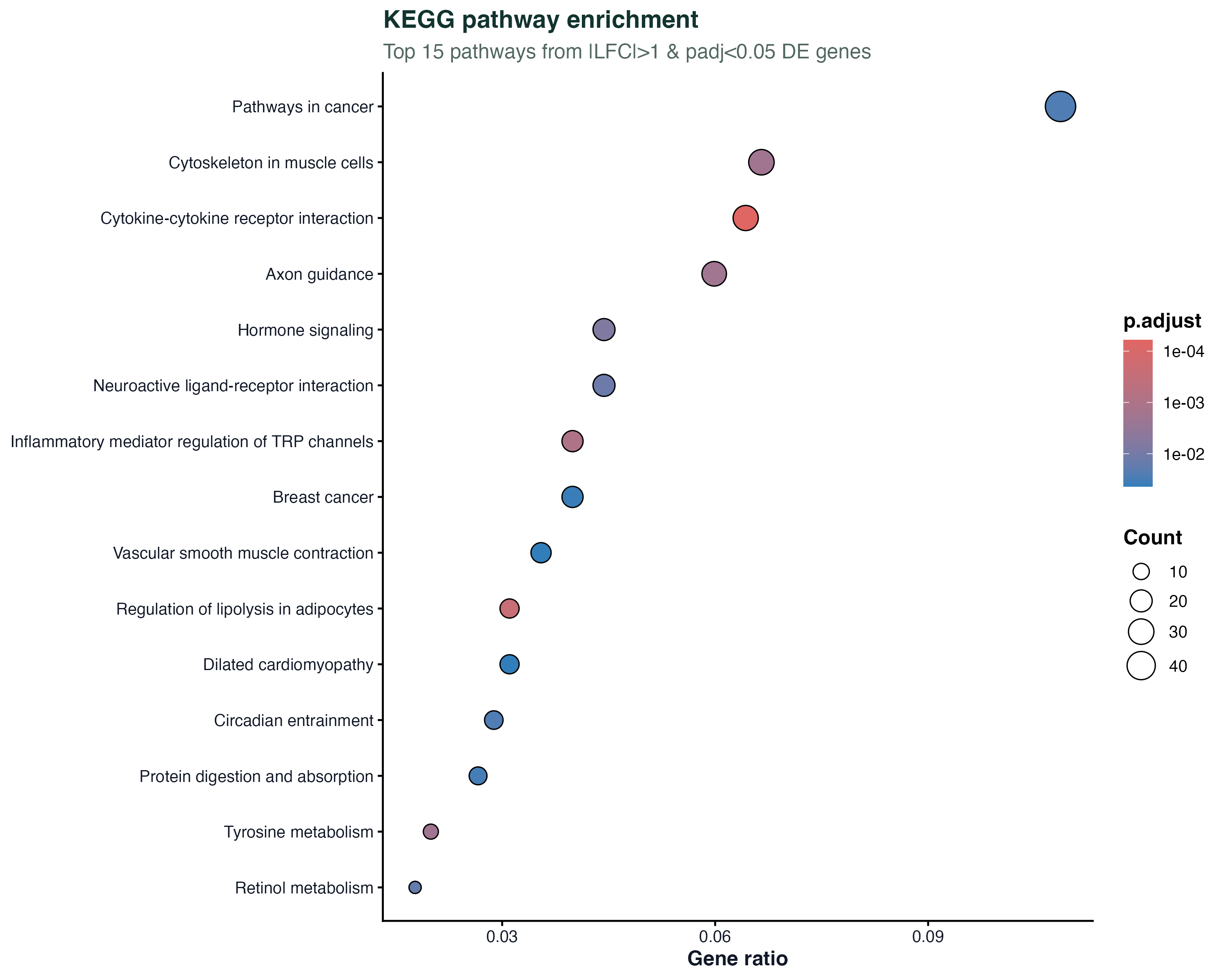
KEGG 比 GO 更贴近"通路"的直觉 —— 每个 pathway 都有一张官方示意图(比如 hsa04110 是 cell cycle),非常适合写报告和做插图。enrichKEGG 默认在线查 KEGG 的 REST API,所以这一步需要能访问 rest.kegg.jp。
注意 KEGG 的 pathway ID 前缀是物种代号(hsa 是人),做小鼠要换成 mmu。
图 5:GSEA top 通路(瀑布图)
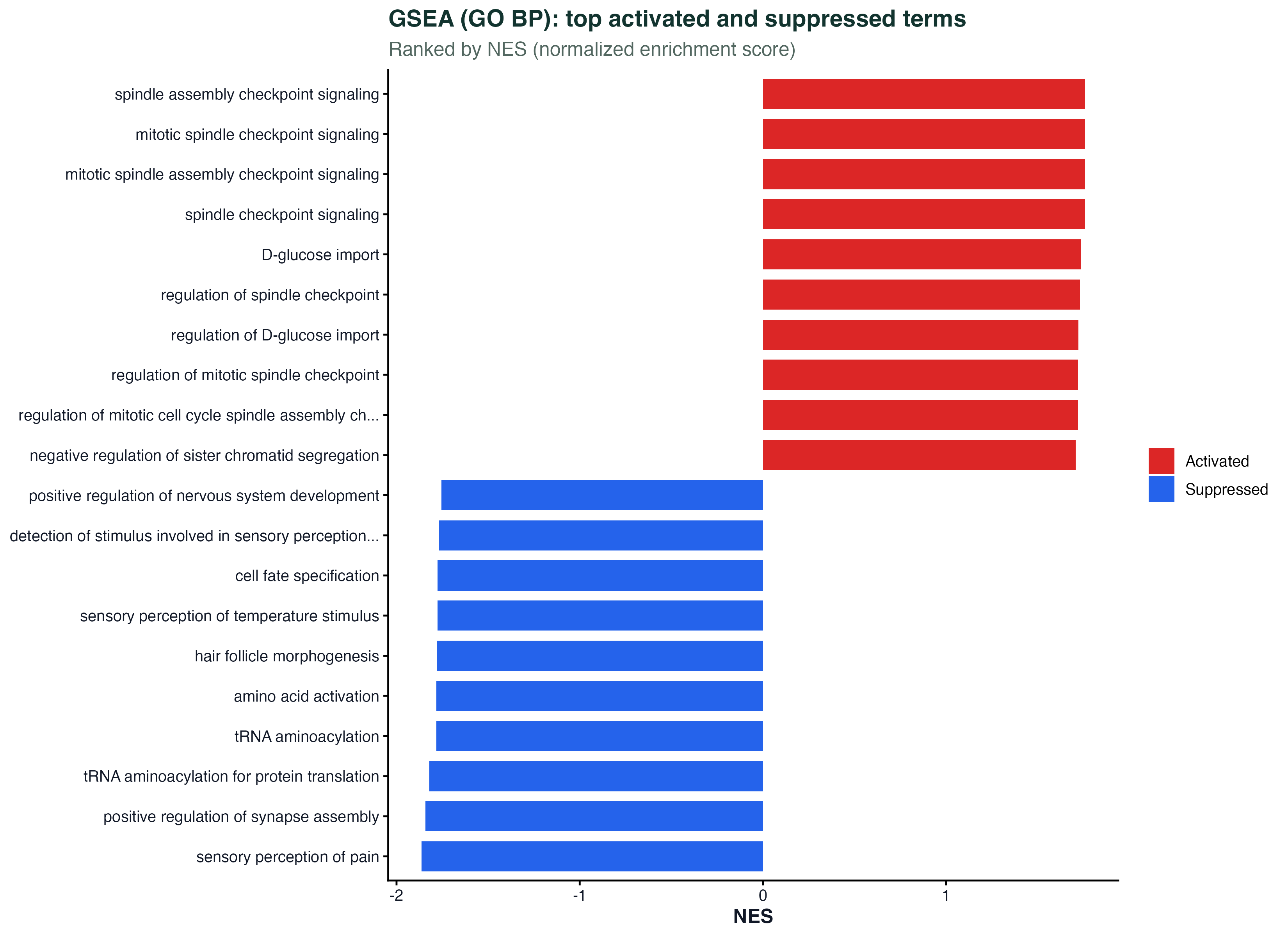
GSEA 按 NES(normalized enrichment score)给每个 term 一个"整体方向性"的打分。红色(上调富集)表示这条通路的基因整�体在处理组里往上走;蓝色(下调富集)表示整体往下。
对 airway,可以看到糖皮质激素相关的响应通路在正方向显著富集,和 ORA 的 dotplot 互相验证。
图 6:GSEA running-score 图
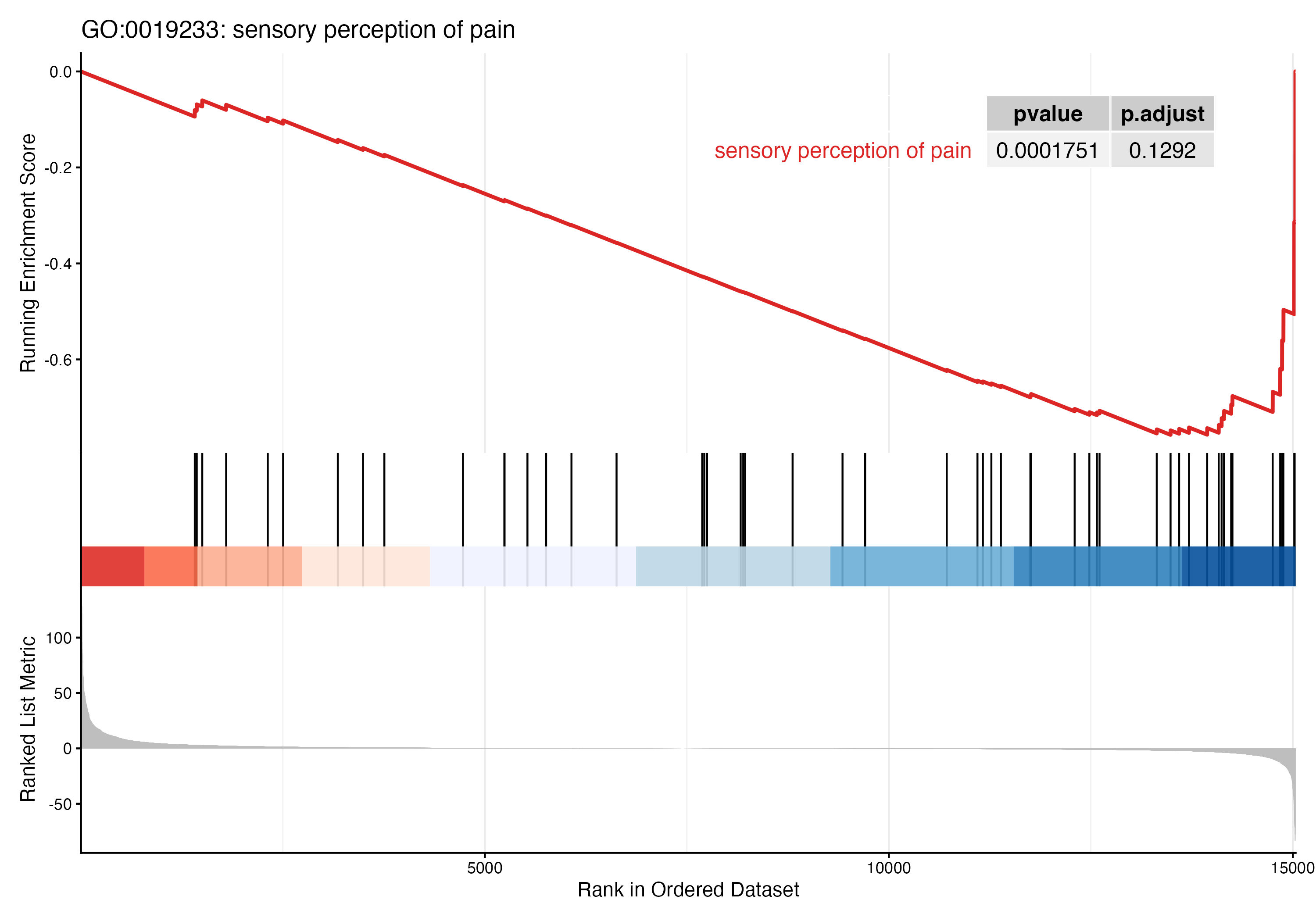
对最强 NES 的那个 term 画的经典 GSEA 图。上半部分的曲线是"在排序里走到这个基因时累计的 enrichment score",峰值越偏离 0 越显著;中间条形表示每次击中一个该通路里的基因时的位置;下半部分是原始 ranking score 的分布。
这张图最核心的是峰值出现在排序的哪一端。峰在左边(高排名那端)= 通路整体上调;峰在右边 = 整体下调;峰在中间没有倾向 = 这条通路不显著。
套到自己数据上
脚本的入口是 res 对象(DESeq2::results() 返回),从其他工具(edgeR 的 glmQLFTest、limma 的 topTable)得到的差异表只要有 log2FoldChange / pvalue / padj 这些列也一样跑。
几个需要根据实际调的参数:
- 物种:
org.Hs.eg.db换成自己物种的 OrgDb(小鼠是org.Mm.eg.db),enrichKEGG(organism = "mmu") - 阈值:默认
padj < 0.05 & |LFC| > 1比较严。小项目或效应较弱的数据可以放宽到padj < 0.1 & |LFC| > 0.58 - 基因集规模:
minGSSize = 15/maxGSSize = 500过滤极端 term,数据小的时候可以降下界 - GSEA 排序统计量:这里用的是
sign(LFC) * -log10(pvalue);也有人用stat(Wald)或log2FoldChange * -log10(padj)
MSigDB 和 fgsea
clusterProfiler::gseGO 和 enrichGO 的基因集只来自 GO。要用 MSigDB 的 Hallmarks、C2(canonical pathways)、C7(immune signatures)这些非常实用的集合,最稳的搭配是 fgsea + msigdbr:
library(msigdbr)
library(fgsea)
# Hallmarks
hallmarks <- msigdbr(species = "Homo sapiens", collection = "H") |>
split(x = .$entrez_gene, f = .$gs_name)
fgsea_res <- fgsea(pathways = hallmarks, stats = rank_vec, minSize = 15, maxSize = 500)
注意 msigdbr 26.x 把基因集改成运行时从 zenodo 下载。国内直连 zenodo 经常慢或失败,可以手动下载一份 msigdb.v2024.1.Hs.json.gz 放到本地,或用 msigdbr::msigdbr_download_db() 指定镜像。
常见坑
坑 1:用 SYMBOL 输入 enrichKEGG
enrichKEGG 期望 ENTREZID。直接喂 SYMBOL 会跑得动但结果几乎全空(命中率 < 5%)—— 因为 KEGG 内部用的是 ENTREZID。永远先 bitr() 转一道。
坑 2:把 background 当成"全部基因"
ORA 的 universe(背景集)应该是这次实验里所有被检测到的基因,不是物种全部基因。设错背景会让显著性虚高(小池子里抓鱼当然显著)。enrichGO(universe = rownames(dds)) 显式传,不要省。
坑 3:GSEA 的 ranking 用了 padj 而不是 LFC
-log10(padj) 都是正值,没有方向,GSEA 跑出来的 enrichment 就只有"越早出现越富集"的方向,看不出激活/抑制。正确做法是 sign(LFC) * -log10(pvalue) 或直接 stat —— 必须有正负。
坑 4:富集结果不去冗余直接报
GO 出来的 top 30 可能只代表 5 个独立的生物学模块,剩下都是冗余 term。emapplot / clusterProfiler::simplify() 去冗余后再写报告。读者看到 30 条很相似的 term 会觉得在凑数。
坑 5:KEGG 在线 API 时不时挂
enrichKEGG(use_internal_data = FALSE) 默认在线查 rest.kegg.jp,有时国内访问慢或断流。长期项目缓存一份本地 KEGG.db 或者用 download_KEGG() 离线,关键分析不要依赖外部 API 当场可用。
下载资源
不想本地装环境?在 BioF3 上跑
GO/KEGG ORA 和 GSEA 各有独立的在线工具,都支持从 DESeq2 结果��一键导入。
下一步
接着深入:
- 04 火山图、热图与富集可视化 — 富集结果出来后怎么做成可发表的 6 张图
横向延伸:
- 02 DESeq2 差异表达分析 — 富集结果质量取决于 DE 表质量,回头检查 DE 是否做对了
- Subramanian 2005 GSEA 原始论文 — GSEA 算法的方法论起点
- clusterProfiler book — 富集分析的全套工具,超出本章范围的工具都在这里
参考资源
离线资料下载
手册 HTML / PDF 已在后台预生成,点击后直接下载�网站静态资源。