07 多工具对比:DESeq2 / edgeR / limma-voom
bulk RNA-seq 差异分析有三个"标配"工具 —— DESeq2、edgeR、limma-voom。它们建模思路不同,但在绝大多数真实数据上结果高度一致。本章做一次 head-to-head 对比:同一份 airway 数据、同一条 design、同一个对比,跑三种工具,看结果是不是真的如传说那样一致。
三种工具的建模差异
| 工具 | 模型 | 特点 |
|---|---|---|
| DESeq2 | 负二项 GLM + 共享离散度估计 + Wald / LRT | 默认的归一化(median of ratios)稳;LFC shrinkage 是一大卖点 |
| edgeR | 负二项 GLM + 经验贝叶斯估计离散度 | TMM 归一化,glmQLFTest 的 F 检验统计学上最稳 |
| limma-voom | 先 voom 把 counts 变成 log-CPM + 权重,再走 limma(线性模型 + 经验贝叶斯) | 速度最快,适合大规模比较;对样本量大的数据尤其稳 |
几个经验:
- 样本量 < 10 对 10,差异不大,三者谁都能用
- 样本量 > 50,limma-voom 的速度优势很明显
- 要做复杂的 contrast(比如多因子设计),limma 的接口最灵活
- 要做单细胞数据的 pseudobulk DE,目前最主流的搭配是 edgeR
为什么 bulk RNA-seq 没有"最佳工具"
很多新手希望有一个"最准的"工具能一锤定音。这不存在,原因是:
1. 三种工具数学上接近等价
DESeq2 和 edgeR 都建模负二项分布,只是离散度估计的先验不同;limma-voom 走线性模型路线,但 voom 的权重设计让它在 RNA-seq 上几乎等价。真实数据上 80%+ 的 DE 基因被三种工具都识别。
2. 各自有各自的最佳场景
- DESeq2 的 LFC shrinkage 在做下游可视化和 GSEA 排序时最稳
- edgeR 的 QLF 检验在 FDR 控制上最严格
- limma-voom 在 100+ 样本规模下速度优势是数量级的
3. 真正影响结果的是上游 QC 和 design
design = ~ condition vs design = ~ batch + condition 带来的差异,远大于"DESeq2 vs edgeR"的差异。先把 QC、批次、设计公式想清楚,工具选哪个其实不那么重要。
实务建议:新项目用 DESeq2(社区最大、教程最多);样本量 > 50 用 limma-voom;做单细胞 pseudobulk 用 edgeR。一线发表都接受。
三条流水线怎么写
DESeq2、edgeR、limma-voom 的 API 有些差别,但核心都是"声明 design + 拟合模型 + 提取 contrast":
# DESeq2
dds <- DESeqDataSetFromMatrix(cnt, colData, design = ~ cell + dex)
dds <- DESeq(dds)
res_deseq <- results(dds, contrast = c("dex", "trt", "untrt"))
# edgeR
dge <- DGEList(cnt) |> calcNormFactors(method = "TMM")
design <- model.matrix(~ cell + dex, data = coldata)
dge <- estimateDisp(dge, design)
fit_edg <- glmQLFit(dge, design)
qlf <- glmQLFTest(fit_edg, coef = "dextrt")
tt_edg <- topTags(qlf, n = Inf)$table
# limma-voom
v <- voom(dge, design, plot = FALSE)
fit_v <- lmFit(v, design) |> eBayes()
tt_lim <- topTable(fit_v, coef = "dextrt", number = Inf)
这三段都用同一份 cnt counts 矩阵和 coldata 样本表。关键在让设计矩阵一致:都是 ~ cell + dex,都检验 dex 的 trt vs untrt 效应。
真实示例:airway 上的三工具对比
配套脚本 bulk07_tools_sci.R 在同一份 airway 数据上跑 DESeq2 / edgeR / limma-voom,做完后对比 6 个维度:
Rscript scripts/bulk07_tools_sci.R
图 1:每个工具的 p 值分布
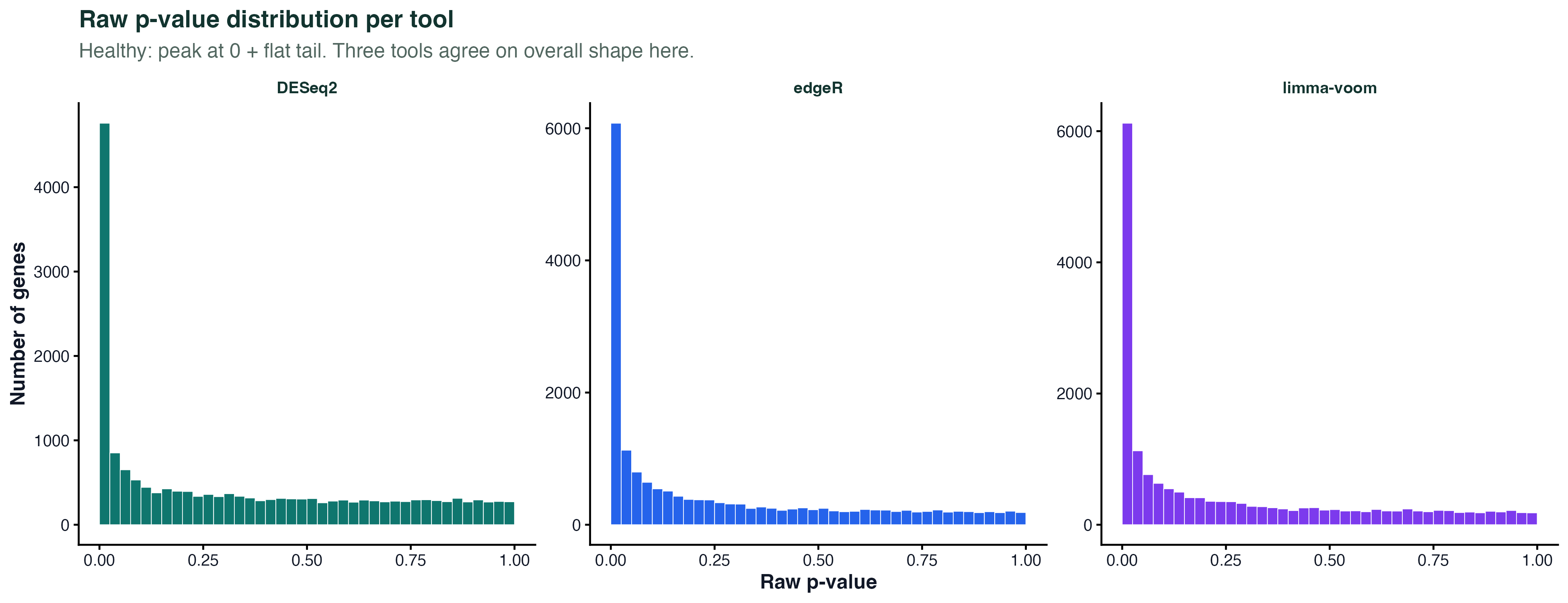
三个工具各自的原始 p 值分布。健康的直方图都有"0 附近 peak + 剩余均匀"的形状。如果某个工具的直方图出现 U 型或者整体左偏,说明它在这份数据上�模型拟合不好。airway 是个"好数据",三个工具都给出了教科书级的 p 值分布。
图 2:不同 padj 阈值下的 DE 基因数
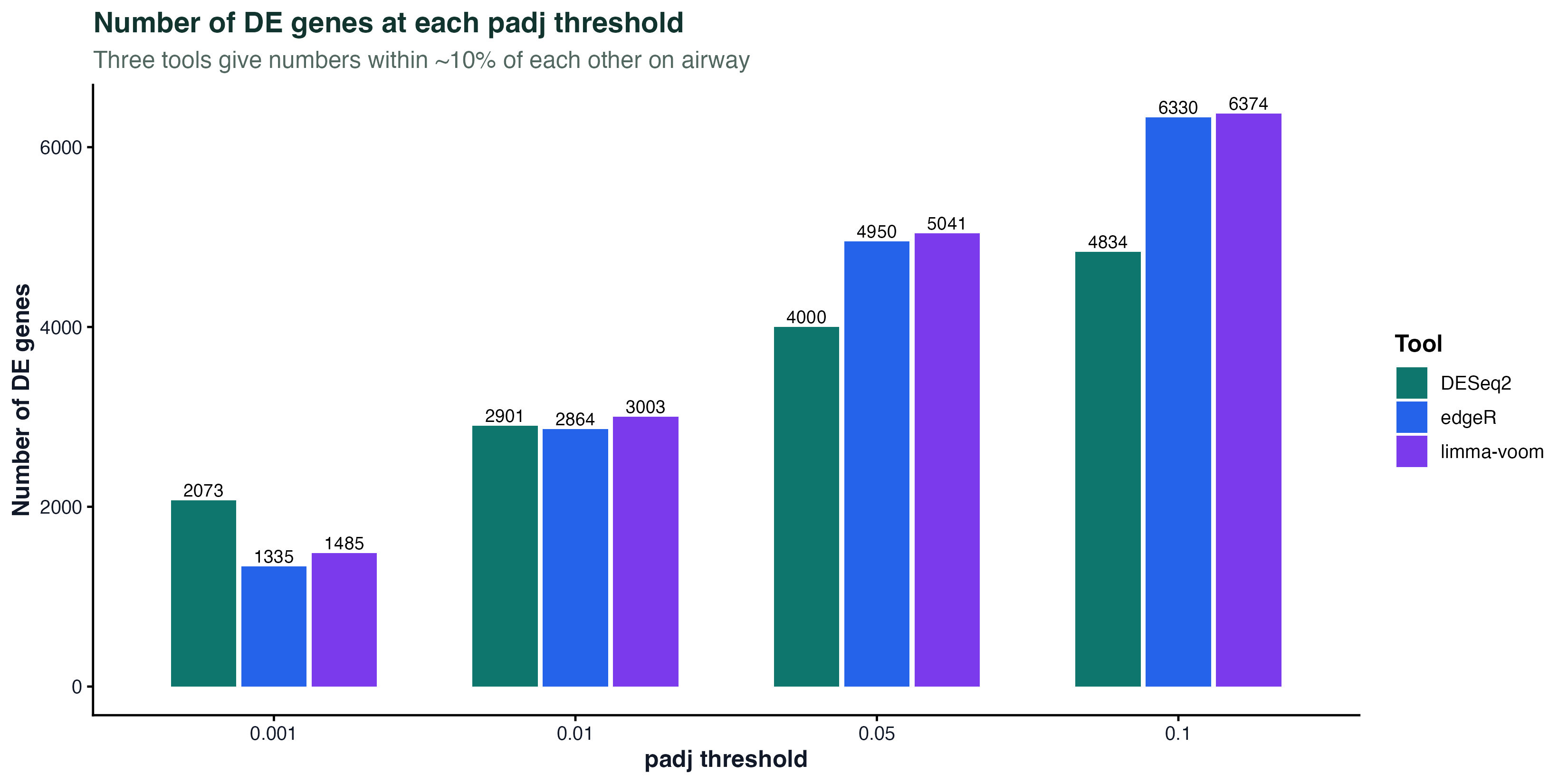
同一个对比,三种工具在 padj < 0.001 / 0.01 / 0.05 / 0.1 四个阈值上的显著基因数。数值差距通常在 10% 以内 —— 这就是"三个工具大差不差"的具体含义。
值得注意的是 edgeR 的 QLF 检验(glmQLFTest)在统计学上是比 DESeq2 的 Wald 更严格的检验,所以 edgeR 的 DE 数经常比 DESeq2 略少。这不是 edgeR 少找到,而是它更严谨。
图 3:DE 基因集的重叠
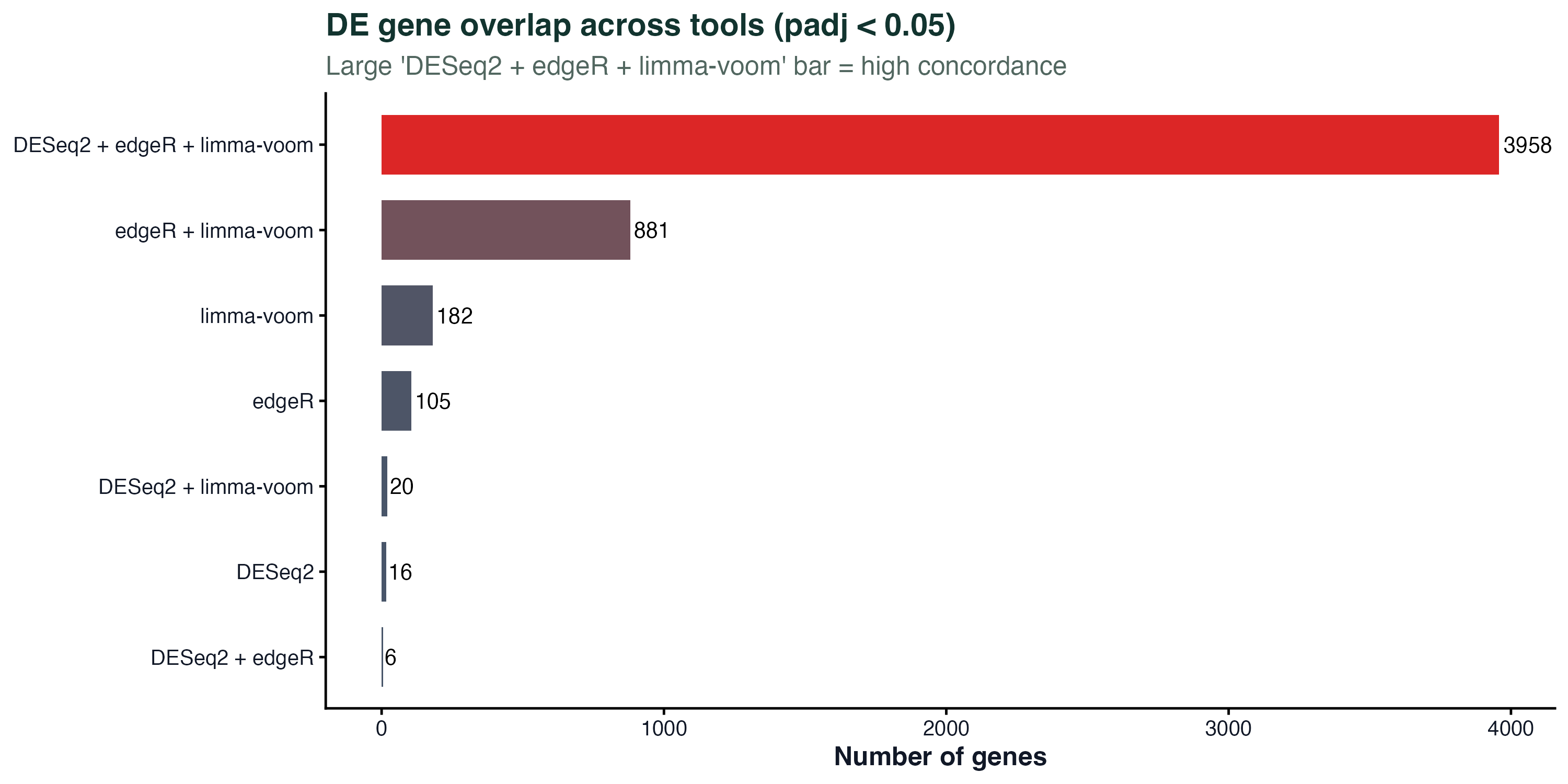
三个工具的 DE 基因集(padj < 0.05)的重叠分布。最大的那一条"DESeq2 + edgeR + limma-voom"说明绝大多数 DE 基因被三个工具同时发现。只被某一个工具独有发现的基因数相对很少,通常是边界基因(接近阈值)。
实用经验:三个工具都说是 DE 的基因,大概率是真�的信号;只被一个工具说是 DE 的,要多看一眼是不是 QC 问题或边界效应。
图 4:log2FC 的散点对比
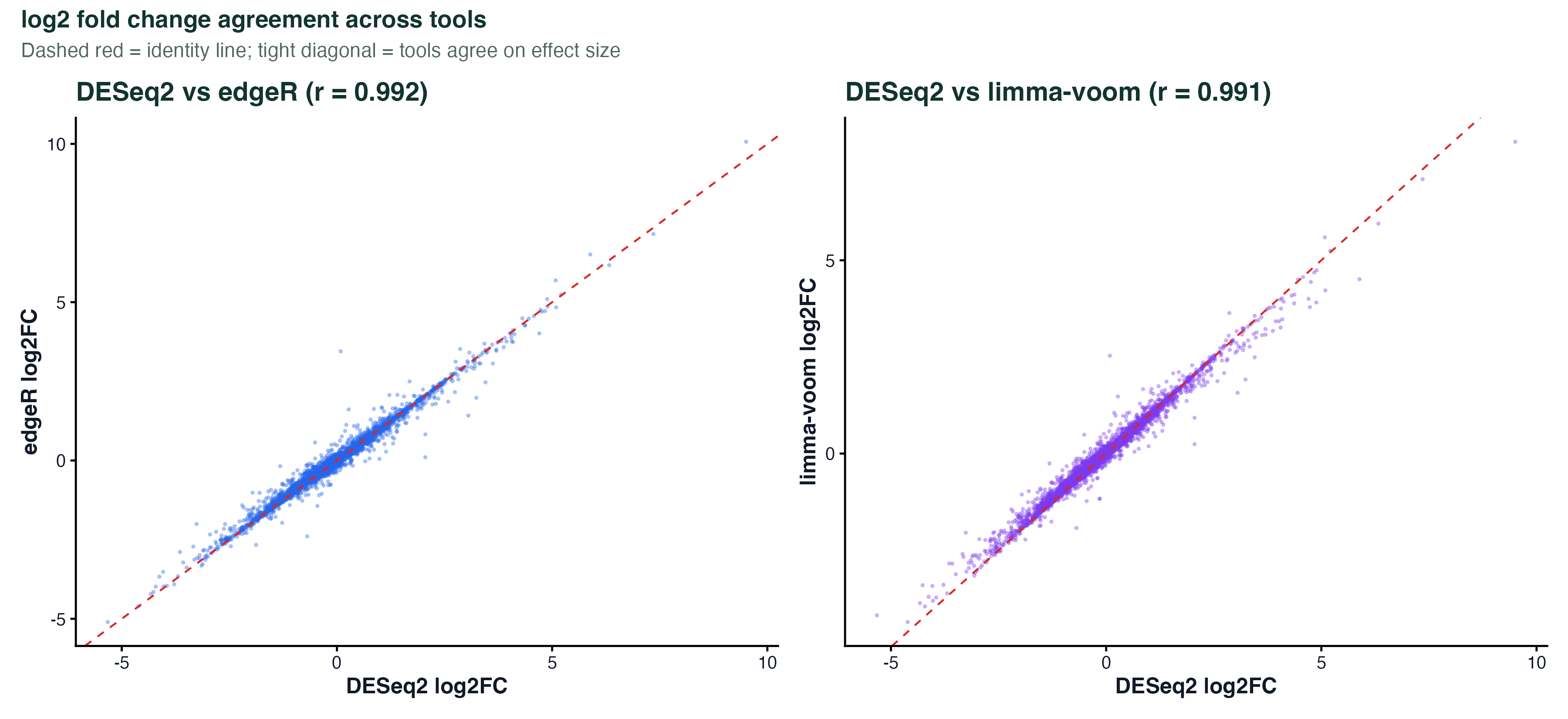
三个工具估计的 log2 fold change 两两散点,红色虚线是 y = x 的对角线。点紧贴对角线 = 三个工具对效应大小的估计几乎一致。
DESeq2 vs edgeR 的相关系数通常在 0.99 左右;DESeq2 vs limma-voom 略低一点(limma-voom 的 LFC 在低表达区会稍微更平),但整体仍 > 0.98。
图 5:p 值的 Spearman 排序相关
![]()
三个工具两两之间,对所有基因按 p 值排序的 Spearman 相关。三个相关系数都 > 0.95。意思是:如果你把"基因按显著性从高到低排名"当作输出(比如用来做 GSEA),三个工具给出的排名几乎完全一样。
这个指标比"DE 基因集重叠"更稳 —— 因为后者会被阈值人为影响,而排序相关和阈值无关。
图 6:工具独有基因的效应大小
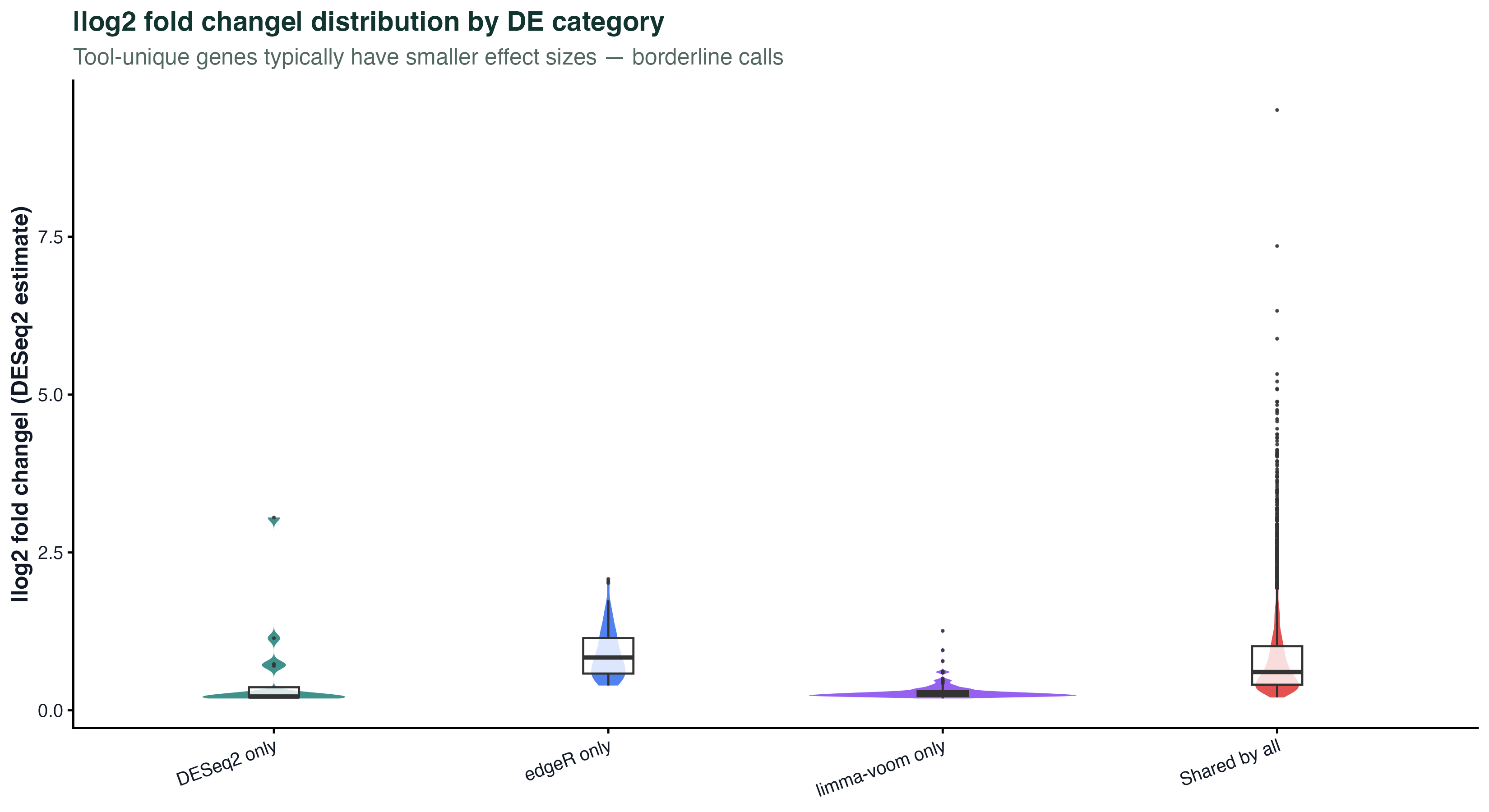
最后一个角度:三种工具独有的那些基因有什么共同点?把每类(A only、B only、C only、三个都有)的 |log2FC| 画成小提琴图。
- Shared by all(三个都有):效应大,中位 |LFC| 可能在 1.5 以上 —— 这些是强信号基因
- 某一个工具独有:效应小,大部分 |LFC| 在阈值附近 —— 边界基因
这张图最重要的解读:工具间的分歧主要发生在"勉强显著"的基因上。真正的强信号,三个工具都抓得到。
什么时候选哪个
实用建议(可以背下来):
| 场景 | 推荐 |
|---|---|
| 样本量小(< 6 对 6)、新项目 | DESeq2,默认最稳 |
| 样本量大(> 30 对 30) | limma-voom,速度最快 |
| 要做 ANOVA-like 检验(LRT / 多因子交互) | DESeq2 的 LRT 或 limma 的 contrasts.fit |
| 单细胞 pseudobulk | edgeR,目前最成熟 |
| 要做严格的 FDR 控制(需要 FPR < 5%) | edgeR QLF,最保守 |
| 要做 LFC shrinkage 做下游 | DESeq2 配 apeglm |
都跑一遍是一个好习惯。在一份新数据上,先用三种工具各跑一遍,如果结果相似,报告其中一种;如果结果差很多,说明数据或设计有问题,要回头看 QC。
除了这三个
其他常见选择:
- NOISeq:非参数方法,对非常态分布友好
- sleuth:搭配 kallisto/Salmon 的 bootstrap 结果使用,能同时建模定量不确定性
- BASiCS:有 spike-in 的数据可用
- DREAM(variancePartition):处理复杂混合效应模型(样本重复测量、配对设计)
刚上手时没必要学这些;先把 DESeq2 / edgeR / limma-voom 中一个摸熟。
常见坑
坑 1:design 公式不一致就直接对比
DESeq2 用 ~ cell + dex、edgeR 用 ~ dex,对比出来 DE 基因差很多 — 当然,模型不一样。三工具对比的前提是 design 矩阵完全一致,否则比的是 design 不是工具。
坑 2:不同工具的 LFC 直接对比
DESeq2 的 LFC 是 shrunken 版本,edgeR 的是 raw,limma-voom 是基于 log-CPM 的回归系数。单位和尺度都不完全一样,散点图能看出趋势但绝对值有偏差。要对比结果一致性,更稳的是比较 p 值排序(Spearman correlation)。
坑 3:拿"工具独有的基因"当差异点报道
DESeq2 独有的 50 个基因里,大概率是 padj 在 0.04-0.06 边缘的基因,换个工具阈值偏一点就过不了。不要把工具差异当成"哪个工具更敏感"的证据,那只是阈值附近的随机扰动。
坑 4:limma-voom 跑出 pvalue = 0
eBayes() 的 t 统计量极端时会输出 pvalue = 0(数值下溢)。pvalue < 1e-300 的基因要 cap 到 1e-300 再做下游,否则 -log10(0) = Inf 让画图崩溃。
坑 5:edgeR 跑 LRT 但忘了 estimateGLMTagwiseDisp
旧版 edgeR 教程用 estimateCommonDisp + estimateTagwiseDisp,新版要用 estimateDisp(一步到位)。两套 API 不混用,老教程的代码在新版可能跑不通或报错。
下载资源
下一步
接着深入:
- 08 可复现分析报告与结果交付 — bulk RNA-seq 专栏的最后一篇,怎么把分析交付出去
横向延伸:
- 02 DESeq2 差异表达分析 — 如果某个工具的结果很奇怪,回头检查 DESeq2 的 QC 图
- Soneson 2018 RNA-seq 工具基准评测 — Nature Methods 的系统对比
参考资源
离线资料下载
手册 HTML / PDF 已在后台预生成,点击后直接下载网站静态资源。