05 多时间点与 LRT 检验
前几章做的都是"两组比较":对照 vs 处理。真实项目里经常遇到更复杂的设计 —— 一条时间曲线,剂量梯度,多个处理条件 —— 这时候两两 Wald 检验就不够了。DESeq2 给出了两个重要工具:
- 设计公式里加交互项:
~ strain + minute + strain:minute,同时估计主效应和交互 - LRT(似然比检验):检验"完整模型相比简化模型是否显著更好拟合",一次拿到所有交互项的综合 p 值
本章用 fission 数据做演示 —— 裂殖酵母在氧化应激下的时间序列:野生型 vs atf21del 突变体,6 个时间点(0/15/30/60/120/180 min),每个组合 3 个生物学重复,共 36 个样本。
多因子设计的核心问题
时间序列、剂量梯度、多条件实验都属于"多因子设计"。要写对 design 公式,先想清楚两个问题:
1. 你想检验的是主效应、交互效应,还是某个特定对比?
| 问题 | 公式 | 检验方式 |
|---|---|---|
| "时间有影响吗?"(不管菌株) | ~ minute | LRT, reduced = ~ 1 |
| "菌株有影响吗?"(不管时间) | ~ minute + strain | LRT, reduced = ~ minute |
| "菌株对时间响应有影响吗?" | ~ strain + minute + strain:minute | LRT, reduced = ~ strain + minute |
| "30 分钟时菌株差异多大?" | 上面同公式 | Wald on strainmut.minute30 |
2. LRT 还是 Wald?
- Wald:一个 contrast 一个 p 值。"A vs B 在某条件下"
- LRT:一组 contrast 一个综合 p 值。"任何 contrast 是否显著"
LRT 适合"我不知道哪个具体对比显著,但想知道有没有任何不一样"。Wald 适合"我已经知道我要看哪个对比"。
时间序列项目典型流程:先 LRT 筛出"响应基因",再 Wald 锁定具体时间点。本章脚本图 5 就是这个用法。
Wald 检验不够用的时候
DESeq2 的默认 results() 做的是 Wald test,回答"某一个对比(contrast)的效应是否显著"。如果问题是:
- "是否存在任何一个时间点上两个菌株有差异?" — Wald 做不了,要对每一对时间点都检验再合并 p 值,麻烦且不严格
- "药物剂量和基因表达是否有剂量-响应关系?" — Wald 只能看某两个剂量的对比
- "某条通路的变化在治疗组和对照组里是否不同?" — 这就是典型的交互项问题
LRT 的解决办法是:给一个"完整模型"和一个"简化模型",问数据在完整模型下的似然是否显著高于简化模型。如果是,说明完整模型里多出来的那些参数(通常是交互项)承载了真实信号。
具体到 fission:
# full 包含交互项、reduced 不包含
dds <- DESeqDataSet(fission, design = ~ strain + minute + strain:minute)
dds <- DESeq(dds, test = "LRT", reduced = ~ strain + minute)
res_lrt <- results(dds)
res_lrt 里每个基因的 padj 回答的是:这个基因对时间的响应,是否依赖菌株? 答案显著的基因就是"时间响应被 atf21 影响的基因"。
真实示例:fission 时间序列
配套脚本 bulk05_timecourse_sci.R 加载 fission → 建立含交互的设计 ��→ LRT 检验 → 画 6 张图:
Rscript scripts/bulk05_timecourse_sci.R
fission 这份数据在"时间主效应"上信号非常强(细胞受到氧化应激),但 strain:minute 的交互信号比较弱(atf21 对响应的影响不那么戏剧)。实际跑下来 LRT 在 padj < 0.05 下只过了几个基因 —— 这本身就是一个重要的教学点:强交互不是所有生物学问题都存在。
图 1:PCA 看整体结构
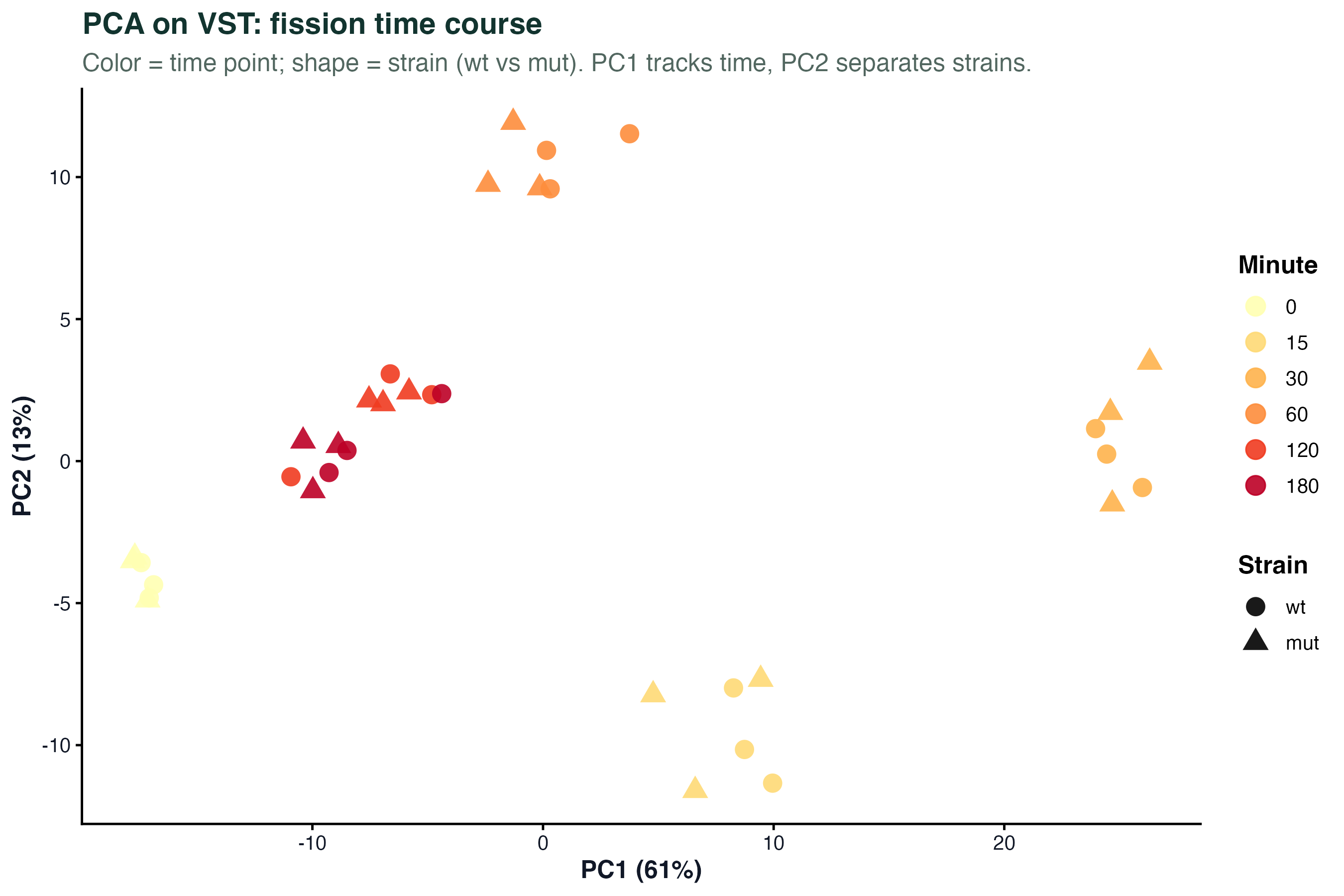
颜色表示时间,形状区分菌株。PC1 几乎就是时间轴 —— 从 0 min(浅色)到 180 min(深红),样本沿着一条轨迹排开。PC2 上两个菌株有一定分离,但比 PC1 上的时间效应弱得多。
这张图回答的第一个问题:时间是最主要的变异来源,说明设计公式里把 minute 当主效应是对的。
图 2:LRT p 值分布与统计量
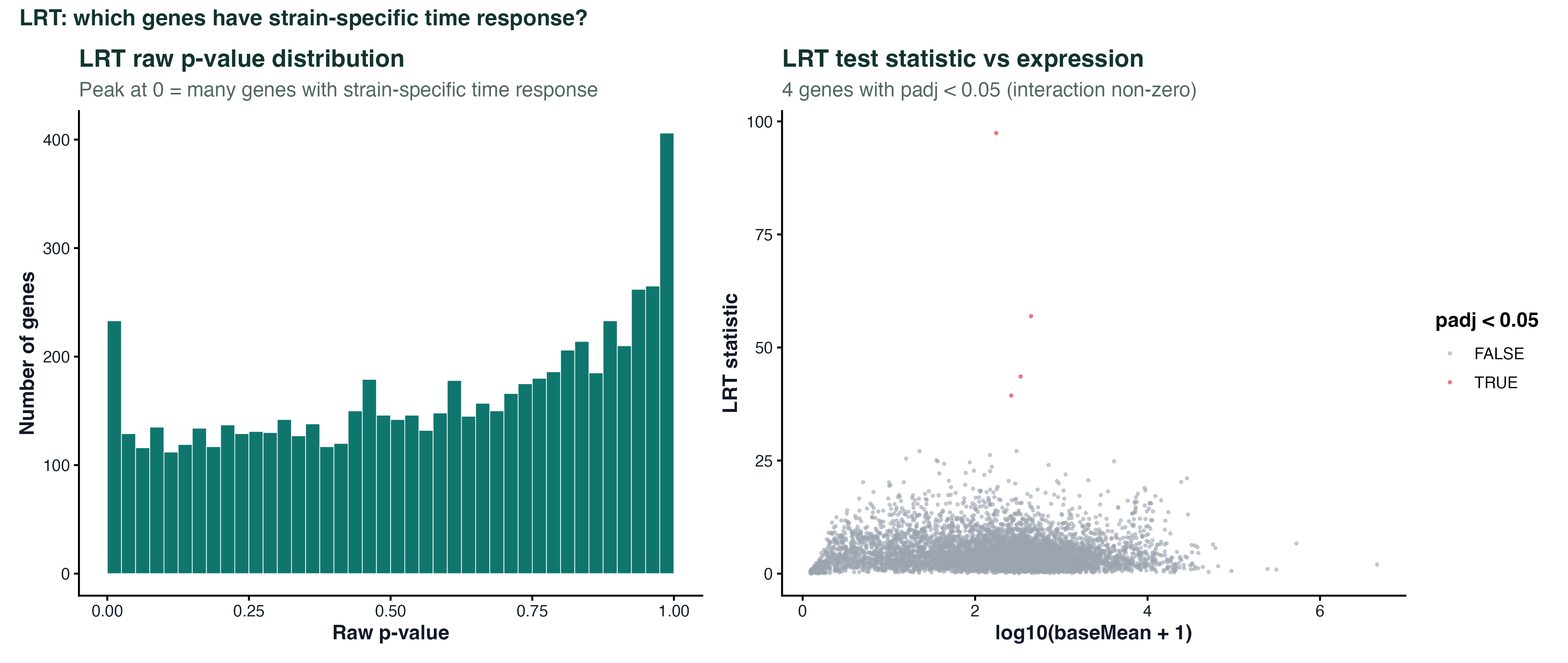
左边是 LRT 原始 p 值的直方图。和 Wald 的直方图一样:0 附近有 peak 说明有真信号,剩余部分接近均匀。 右边横轴是 log10(baseMean),纵轴是 LRT 统计量。LRT 统计量越大说明"完整模型比简化模型好"的程度越强。高表达基因(右侧)通常统计量更大 —— 这是 RNA-seq 的常见现象:高表达基因数据多、功效高。
图 3:Top LRT 基因的时间轨迹
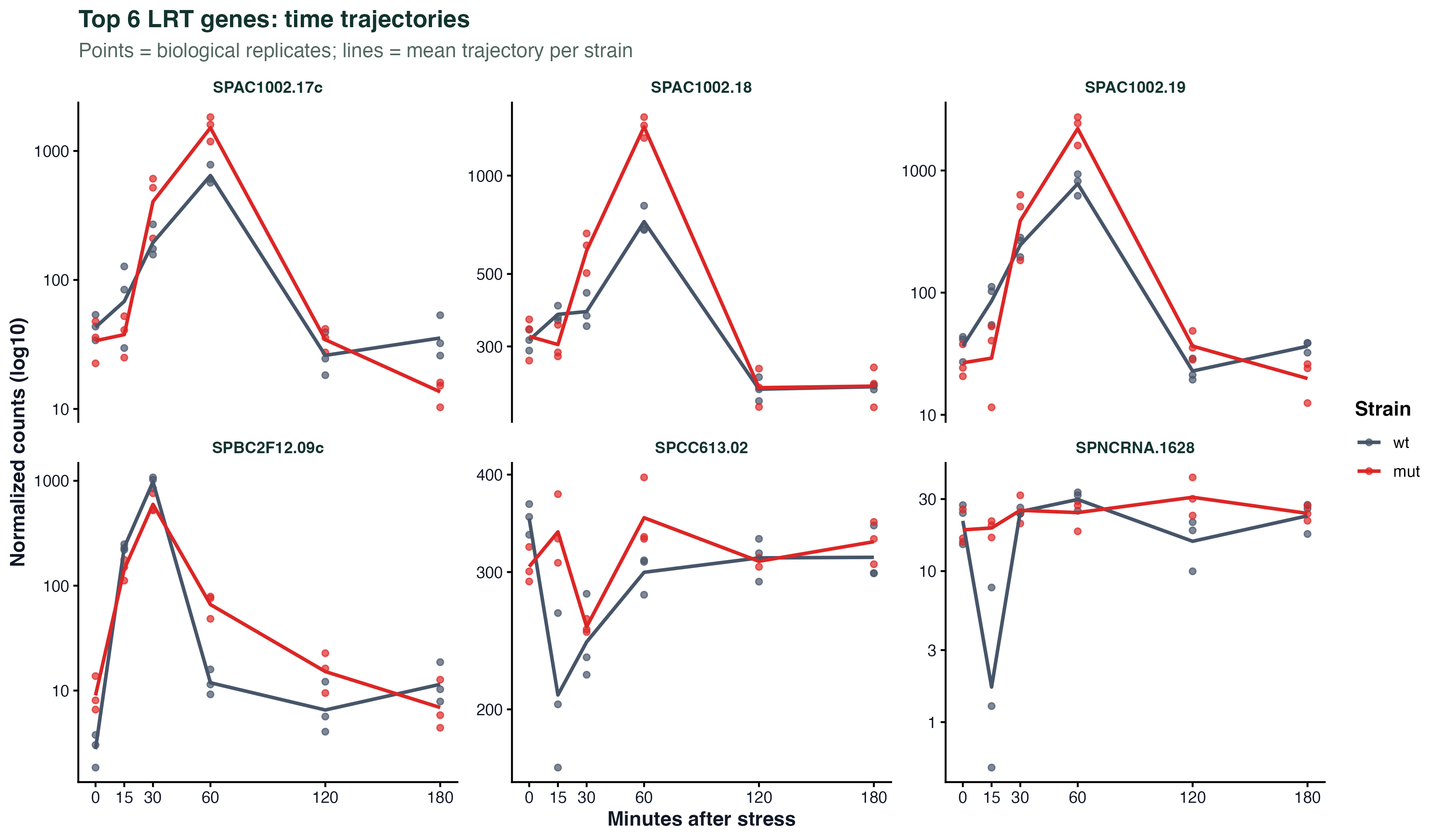
对 padj 最小的 6 个基因,把两个菌株的 normalized counts 画成时间曲线(点 = 重复,粗线 = 同时间点的均值)。可以看到 wt 和 mut 的曲线在某些时间点"分叉" —— 这就是 LRT 捕获的信号:两条曲线的形状不一样。
这种图在写论文时是"讲故事"最核心的图:把抽象的 LRT 统计量具象成"这个基因在 30 分钟的时候开始在突变体里下降"。
图 4:聚类的时间轨迹
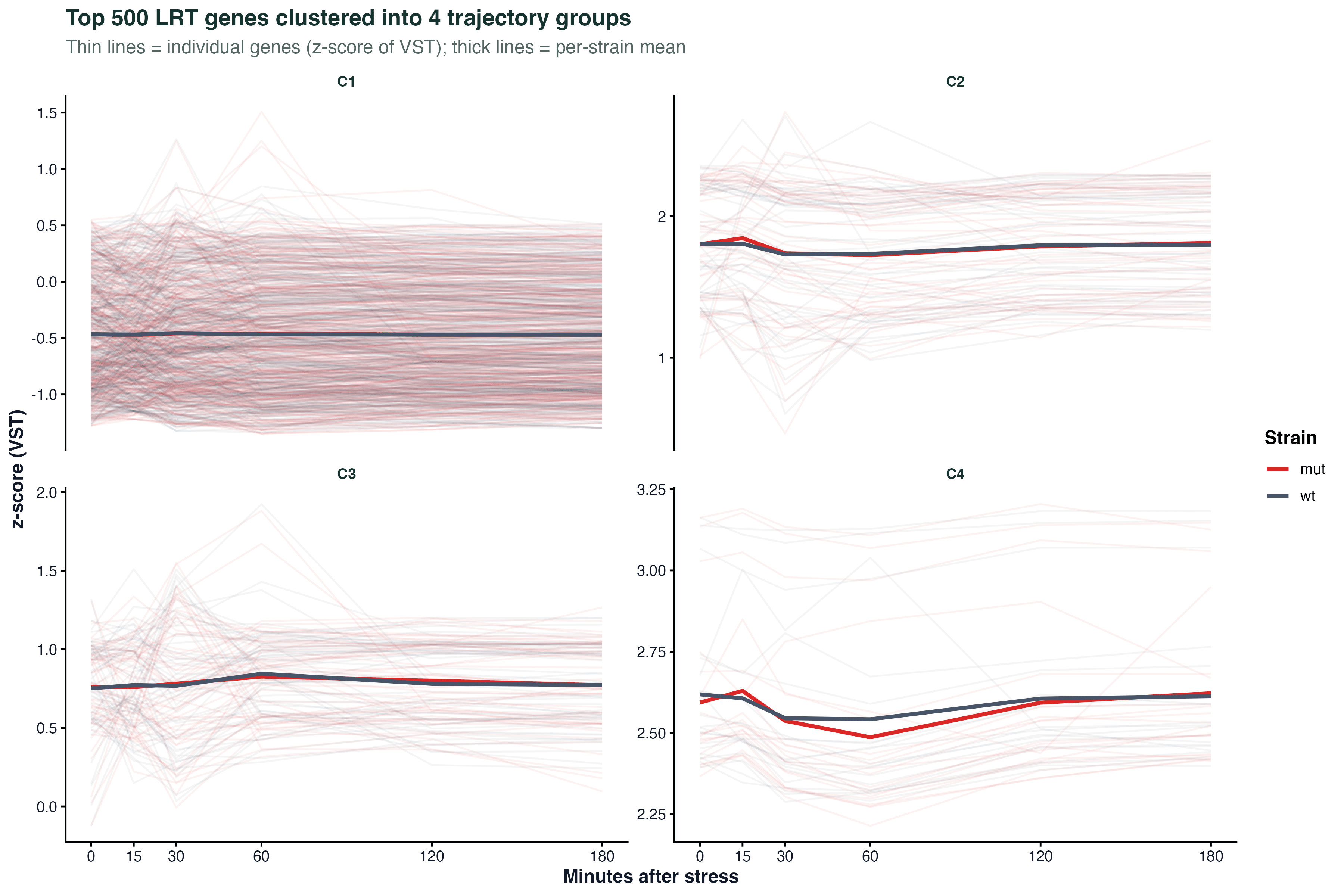
取 LRT 排名前 500 的基因,做一次层次聚类,分 4 组。每组里每个基因画一条半透明细线(z-score),最上面的粗线是两个菌株各自的均值。
这张图的信息量很大:
- 哪些基因"早响应、早下降"(C1)
- 哪些"晚响应、持续升高"(C3)
- 哪些在两个菌株间方向一致但幅度不同(C2)
- 哪些在一个菌株里响应、另一个菌株里不响应(C4)
真实项目里这张图往往比 dotplot 更能直接生成生物学假设。
图 5:特定时间点的交互 volcano
LRT 告诉你"哪些基因的响应依赖菌株",但不告诉你在哪个时间点差异最大。要定位具体时间点,改用 Wald 检验提取单独一个交互项的效应:
dds_wald <- DESeq(dds) # default test = "Wald"
# 查 resultsNames(dds_wald) 看有哪些系数
res30 <- results(dds_wald, name = "strainmut.minute30")
strainmut.minute30 这个系数回答的是:在 30 分钟时,mut 和 wt 之间的差异,相对于时间 0 的基线差异,是否显著? 正值 = mut 在 30 分钟比 wt 升得更多;负值 = mut 升得更少或反而下降。
图 6:Top LRT 基因热图(按时间排序)
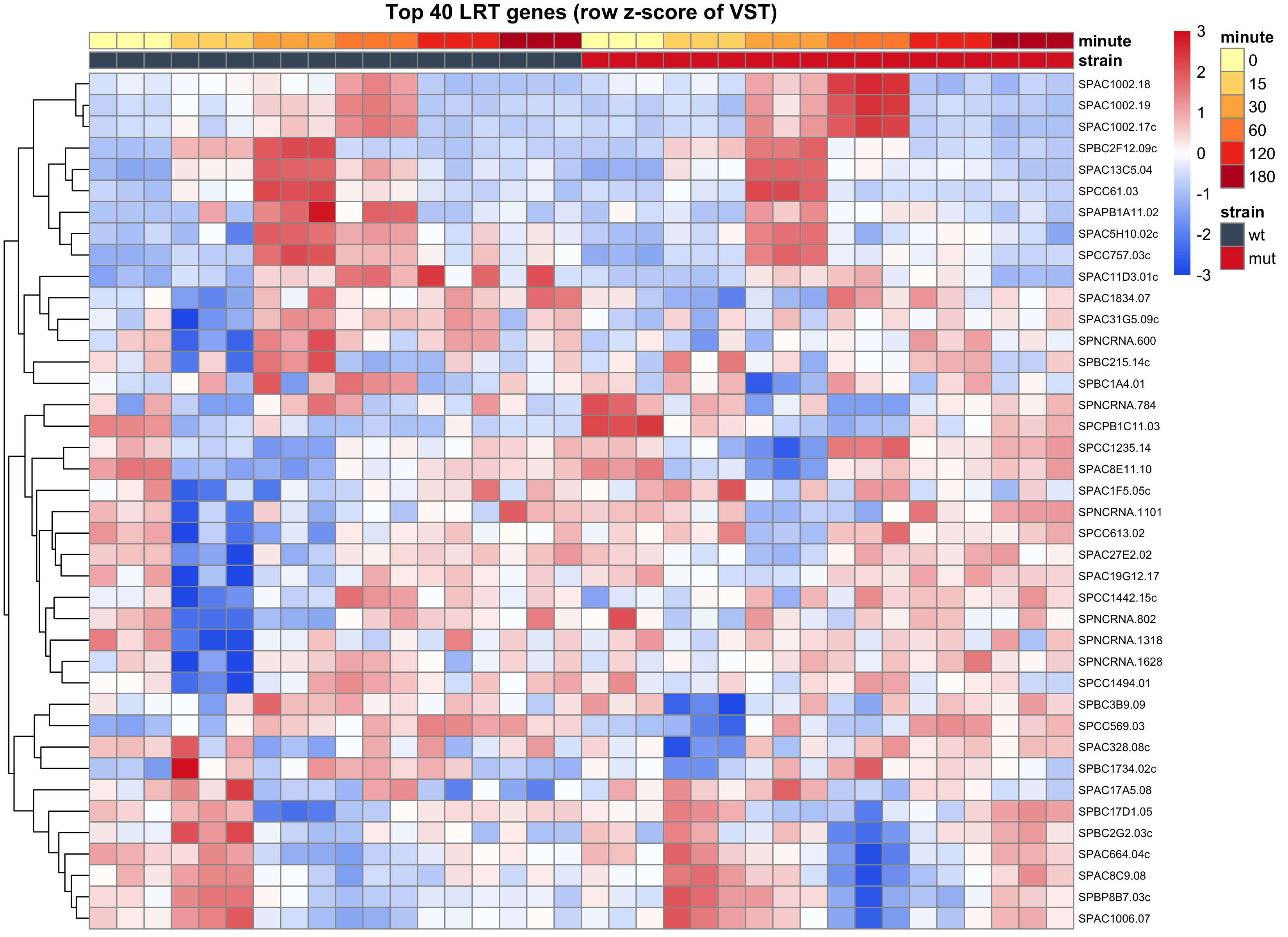
列按菌株 + 时间排序(不做列聚类),行是 top 40 LRT 基因的 row z-score。横向左到右能看到每个基因沿时间的变化;纵向看哪些基因变化模式相似。
其他常见的复杂设计
设计公式的写法对应生物学问题。一些速查:
| 问题 | design | 检验方式 |
|---|---|---|
| 两组比较 | ~ condition | results(dds, contrast = c("condition","B","A")) |
| 控制批次 | ~ batch + condition | 同上 |
| 时间效应 | ~ minute(因子) | LRT: reduced = ~ 1 |
| 时间 × 菌株交互 | ~ strain + minute + strain:minute | LRT: reduced = ~ strain + minute |
| 连续剂量响应 | ~ dose(数值变量) | Wald on dose coefficient |
| 多条件 + 批次 | ~ batch + tissue + condition | results(..., contrast = ...) |
两个关键原则:
- 要检验的变量一般放 design 最后,results() 默认取它
- LRT 时
reduced要和full只差你关心的那些项 —— 差得越多,LRT 就是在同时检验那些项的组合,解读会变乱
和 single-cell 的对比
单细胞里做"轨迹推断 + 沿拟时序的差异"(比如 Slingshot + tradeSeq),思路和 bulk 里 LRT 很像:都在问"基因表达是否沿着某个连续变量系统性变化"。主要差别:
- bulk 的 minute 是实验里设好的时间点,维度离散、样本多;单细胞的 pseudotime 是推断出的连续变量,每个细胞一个值
- bulk 的 LRT 基于 counts 的负二项分布;
tradeSeq用 GAM 拟合连续轨迹
常见坑
坑 1:把时间当数值变量
design = ~ minute(minute 是 numeric)会假设"基因表达沿时间线性变化"。但生物学响应通常不是线性的(先升后降、滞后峰值)。默认把 minute 转成 factor:dds$minute <- factor(dds$minute, levels = c(0, 15, 30, 60, 120, 180)),让模型自己估计每个时间点的水平。
只有在你确实想假设"剂量响应是单调线性"时才把 dose 留作 numeric。
坑 2:reduced 和 full 差太多导致 LRT 解读混乱
LRT 检验的是 "full 比 reduced 多出来那些项的整体显著性"。如果 full = ~ batch + strain + minute + strain:minute、reduced = ~ 1,LRT 同时检验 4 件事的综合显著性 — 结果显著也说不清是哪个驱动。reduced 只比 full 少你关心的那一项,结果才好解读。
坑 3:LRT 之后用 Wald 时忘了重新跑 DESeq
LRT 模式下 dds <- DESeq(dds, test = "LRT", reduced = ...) 拟合的是 LRT 模型;要做 Wald 必须 dds_wald <- DESeq(dds, test = "Wald") 重新跑一遍,不能直接 results(dds, name = ...) 取系数,那会用 LRT 时估的 SE,结果不对。
坑 4:交互项基因数比预期少很多
时间×菌株交互的强信号在很多生物学体系里就是稀疏的 — 大部分基因对时间的响应在两组里其实差不多,只是少数关键基因有差异。fission 这份数据里 LRT padj < 0.05 也只过几十个基因,是正常的,不是分析做错了。先看 PCA 是否有明显的菌株分离判断信号强度。
坑 5:用 vst 后的值做时间轨迹但忽略了 batch
vst(dds, blind = FALSE) 会用 design 信息去除 batch 影响;blind = TRUE 不会。做时间轨迹可视化时务必 blind = FALSE,否则 batch 噪声会把每条轨迹搞乱。
下载资源
下一步
接着深入:
- 06 批次效应 — 时间序列实验经常跨多个测序批次,下一步是把 batch 也加进 design
- 07 多工具对比 — limma 的
contrasts.fit在多因子设计下有时比 DESeq2 LRT 更灵活
横向延伸:
- 单细胞 05 轨迹推断 — bulk 的 LRT 与单细胞的 pseudotime 在思路上互通
- DESeq2 时间序列 vignette — 官方的更详细示例
参考资源
离线资料下载
手册 HTML / PDF 已在后台预生成,点击后直接下载网站静态资源。