module02
速答
Q: DESeq2 为什么用负二项分布而不是泊松? A: 泊松假设均值=方差,RNA-seq 数据存在过度离散(方差 > 均值),负二项分布多一个离散度参数能拟合这种过度离散。
Q: DESeq2 的 padj 和 pvalue 有什么区别? A: padj 是 Benjamini-Hochberg 校正后的 p 值,控制假阳性率。多基因检验时必须用 padj(通常 < 0.05),不能用原始 pvalue。
Q: log2FC 阈值设多少? A: 常用 |log2FC| > 1(即 2 倍变化)。宽松可降到 0.58(1.5 倍),严格可到 1.5(2.8 倍)。结合 padj 一起筛。
Q: LFC shrinkage 是什么,要不要做? A: 对 log2FC 做收缩估计(apeglm / ashr),减少低表达基因的极端 fold change。推荐做,结果更可信、火山图更好看。
Q: DESeq2 和 edgeR 怎么选? A: DESeq2 文档完善、默认参数稳健,适合大多数场景;edgeR 更灵活、速度更快,适合有经验的分析者。结果通常高度一致。
02 DESeq2 差异表达分析
DESeq2 是 bulk RNA-seq 差异分析里最常用的工具。它假设 counts 服从负二项分布,用每个基因的表达均值和离散度估计显著性,加上 Benjamini-Hochberg 校正给出最终的 padj。流程非常规整,稳到几乎是默认选择。
这一章用 DESeq2 官方教程数据 airway 走一遍完整流程。数据是 8 个人类气道上皮细胞样本 —— 4 个对照 + 4 个糖皮质激素(地塞米松,dex)处理,来自 Himes et al. 2014(PLoS ONE 9(6): e99625)。
为什么是负二项分布而不是泊松或正态
DESeq2 模型选择不是随便拍脑袋决定的,理解这一点能让你看懂很多默认行为:
- 不是正态分布:counts 是非负整数,且方差不等于均值(高表达基因的方差比泊松预期的大很多)
- 不是泊松分布:泊松假设方差 = 均值,但真实 RNA-seq 数据 over-dispersed —— 同一条件下生物学重复之间方差远大于均值
- 是负二项分布:在泊松基础上加一个 dispersion 参数,描述"额外的方差"。这个 dispersion 在低表达基因数据少时估不准,DESeq2 用"所有基因共同拟合一条 mean-dispersion 曲线"来稳定估计
这意味着什么:
- counts 矩阵必须是未归一化的整数(raw counts),不能是 TPM / RPKM / 已 log 变换的值
- DESeq2 需要至少 2 个生物学重复才能估 dispersion;3 个起步、4-6 个稳
- 强行用单样本(1 vs 1)跑 DESeq2 会强制把 dispersion 拉到全局均值,结果不可信
核心流程是三步
DESeq2 的调用界面非常简洁。真正的 API 就三步:
dds <- DESeqDataSet(se, design = ~ batch + condition)
dds <- DESeq(dds)
res <- results(dds, contrast = c("condition", "trt", "untrt"))
- 构建
DESeqDataSet对象,声明设计公式(design最后一列是要检验的变量) DESeq()一个调用里做完归一化、离散度估计、Wald 检验results()提取指定对比的差异结果
真实项目的工作重点不在这三行代码上,而在围绕它的 QC 和解释:样本是否正常、模型是否合适、结果是否可信。下面按这个顺序走。
加载 airway 并构建 DESeq 对象
library(DESeq2)
library(airway)
data(airway)
se <- airway
# 把 untrt 设为参考水平,这样 log2FC 是 "trt 相对 untrt"
se$dex <- relevel(se$dex, ref = "untrt")
# design: 控制细胞系差异,检验 dex 处理效应
dds <- DESeqDataSet(se, design = ~ cell + dex)
# 预过滤:counts 总和 < 10 的基因直接丢掉
# 不影响统计正确性,但能加速并减少内存
keep <- rowSums(counts(dds)) >= 10
dds <- dds[keep, ]
airway 的 colData 里 cell 是细胞系(4 个不同来源),dex 是处理变量。design = ~ cell + dex 的意思是"在控制细胞系差异之后看 dex 的影响"。设计公式里变量的顺序无所谓,但要检验的变量一般放最后,results() 默认会拿最后一个变量的第一个对比。
跑 DESeq 并看总体结果
dds <- DESeq(dds)
res <- results(dds, contrast = c("dex", "trt", "untrt"))
summary(res)
summary(res) 会告诉你上下调��基因各有多少个(默认阈值 padj < 0.1)。对 airway 这份数据,通常会看到几千个显著差异基因 —— 糖皮质激素是强效应通路,差异很明显。
真实示例:airway 上的完整 DESeq2 分析
配套脚本 bulk02_deseq2_sci.R 把上面的流程完整跑了一遍,输出 6 张真实数据图和一份 DE 基因表。装好 DESeq2 + airway + apeglm 就能直接跑,不需要下载 FASTQ:
Rscript scripts/bulk02_deseq2_sci.R
脚本里做的事:预过滤 → DESeq() → results() → lfcShrink(apeglm) → 6 张图 → 输出 DE 表。下面逐张看。
图 1:每个样本的库大小
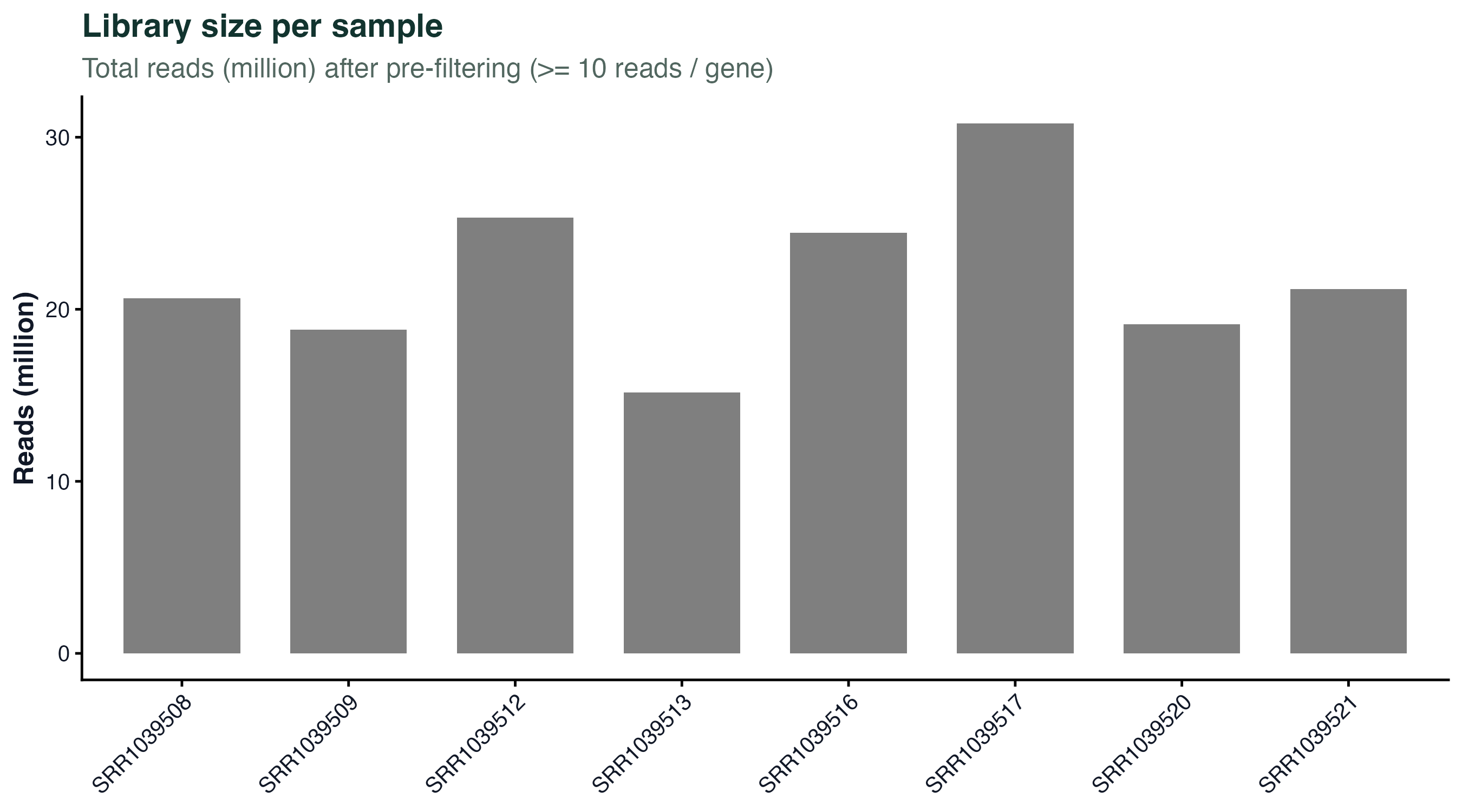
横轴是样本 ID,纵轴是预过滤之后每个样本的总 reads(百万)。这张图是最基础的 QC:库大小应该在一个量级,如果有某个样本只有其他样本的 1/5,就要回去看测序报告。airway 里 8 个样本大小相当,没问题。
图 2:VST 归一化后的 PCA
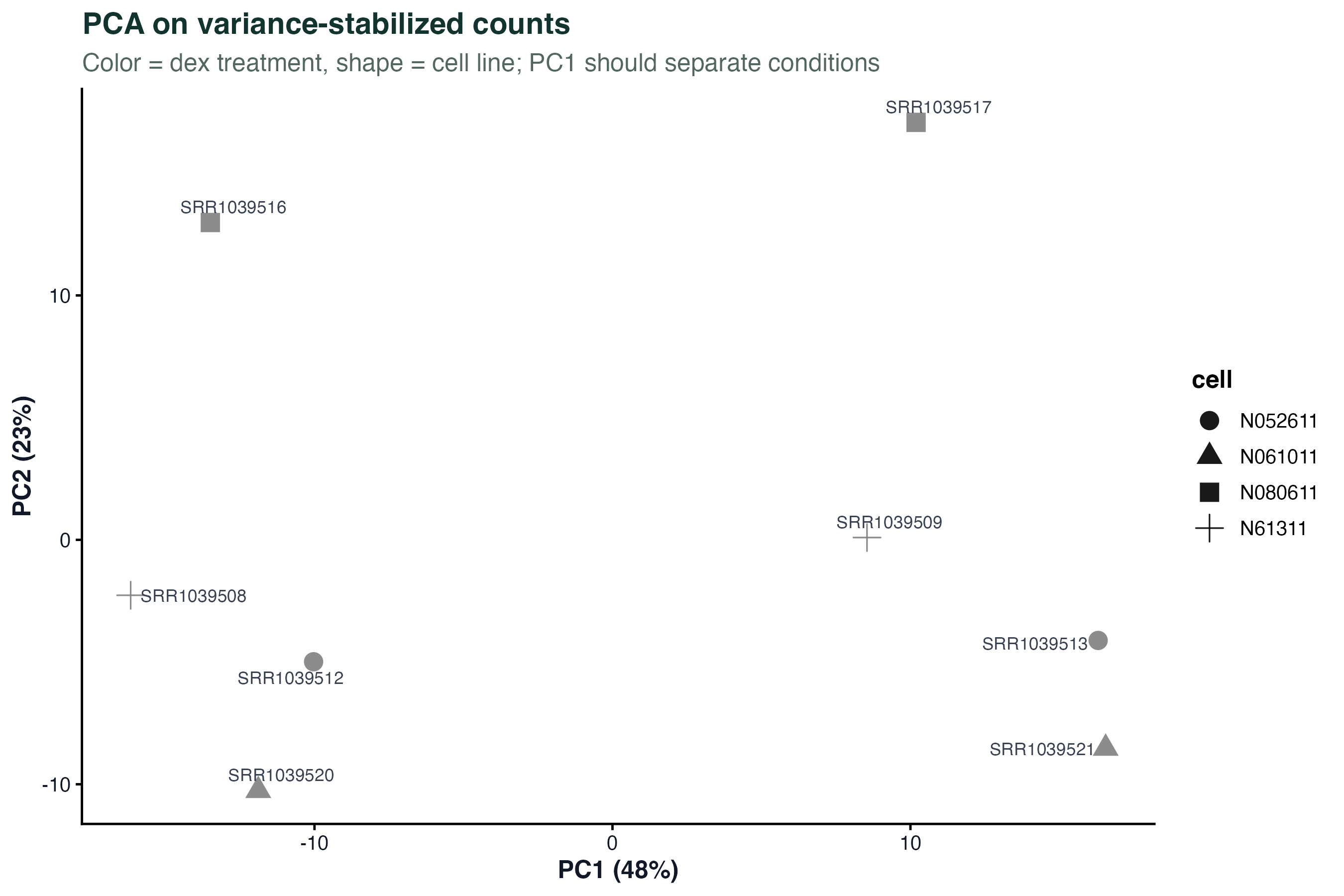
vst(dds, blind = FALSE) 做方差稳定变换 —— 让高表达和低表达基因的方差都差不多,这样 PCA 才不被少数高表达基因主导。颜色是 dex 处理,形状是细胞系。
这张图回答两个问题:
- PC1 是什么:理想情况下 PC1 直接把处理组和对照组分开。airway 里 PC1 正是 dex 轴。
- 细胞系差异大不大:同样颜色但不同形状的点会偏开,说明细胞系间有个体差异。这就是 design 里加
cell的原因。
图 3:样本间欧式距离
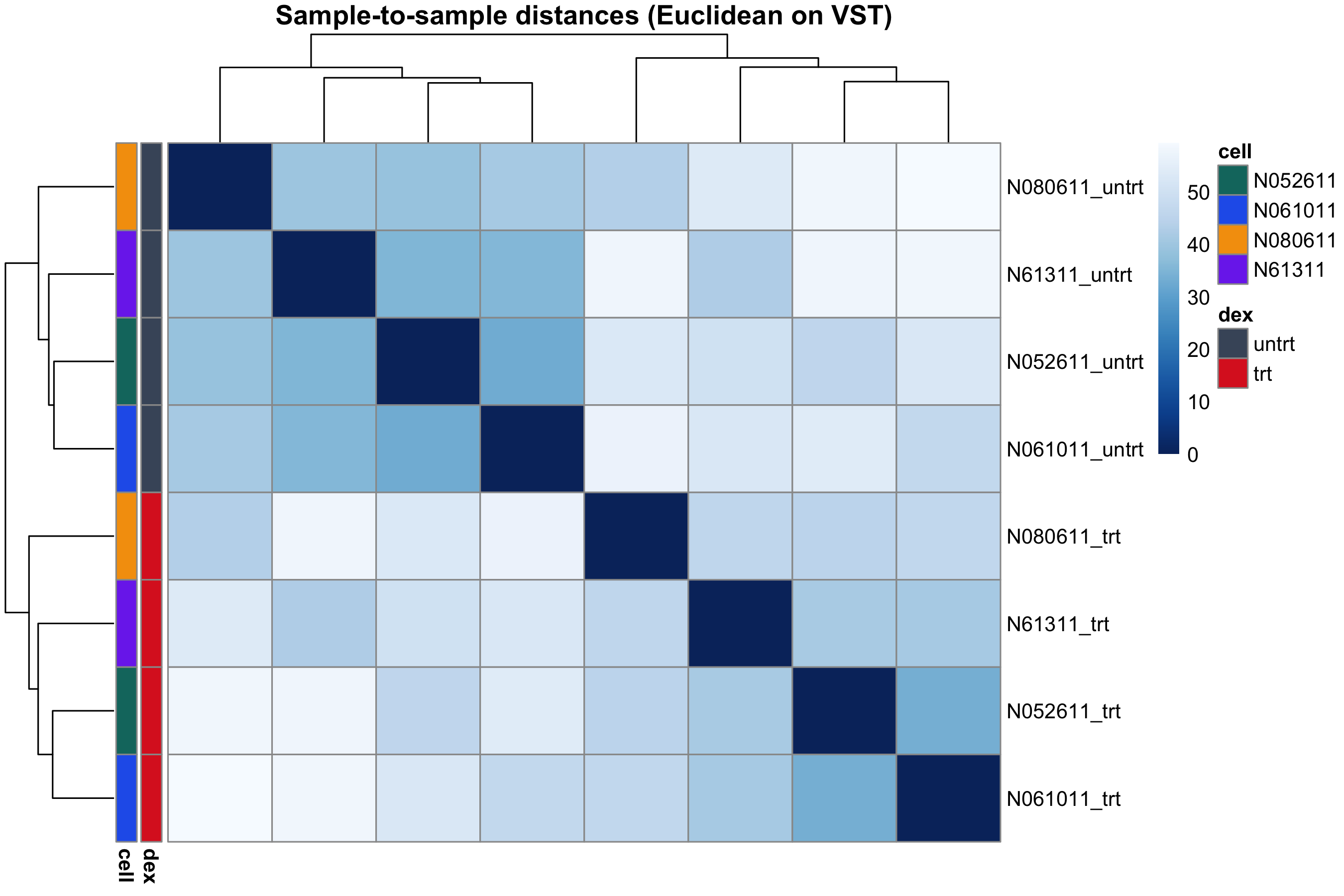
样本两两之间的欧式距离(在 VST 空间)画成热图,再聚一次类。同一条件 + 同一细胞系的样本应该靠在一起。如果某个样本跟同组的样本距离反而大过别组,要小心标签弄错或者该样本有问题。
图 4:离散度估计
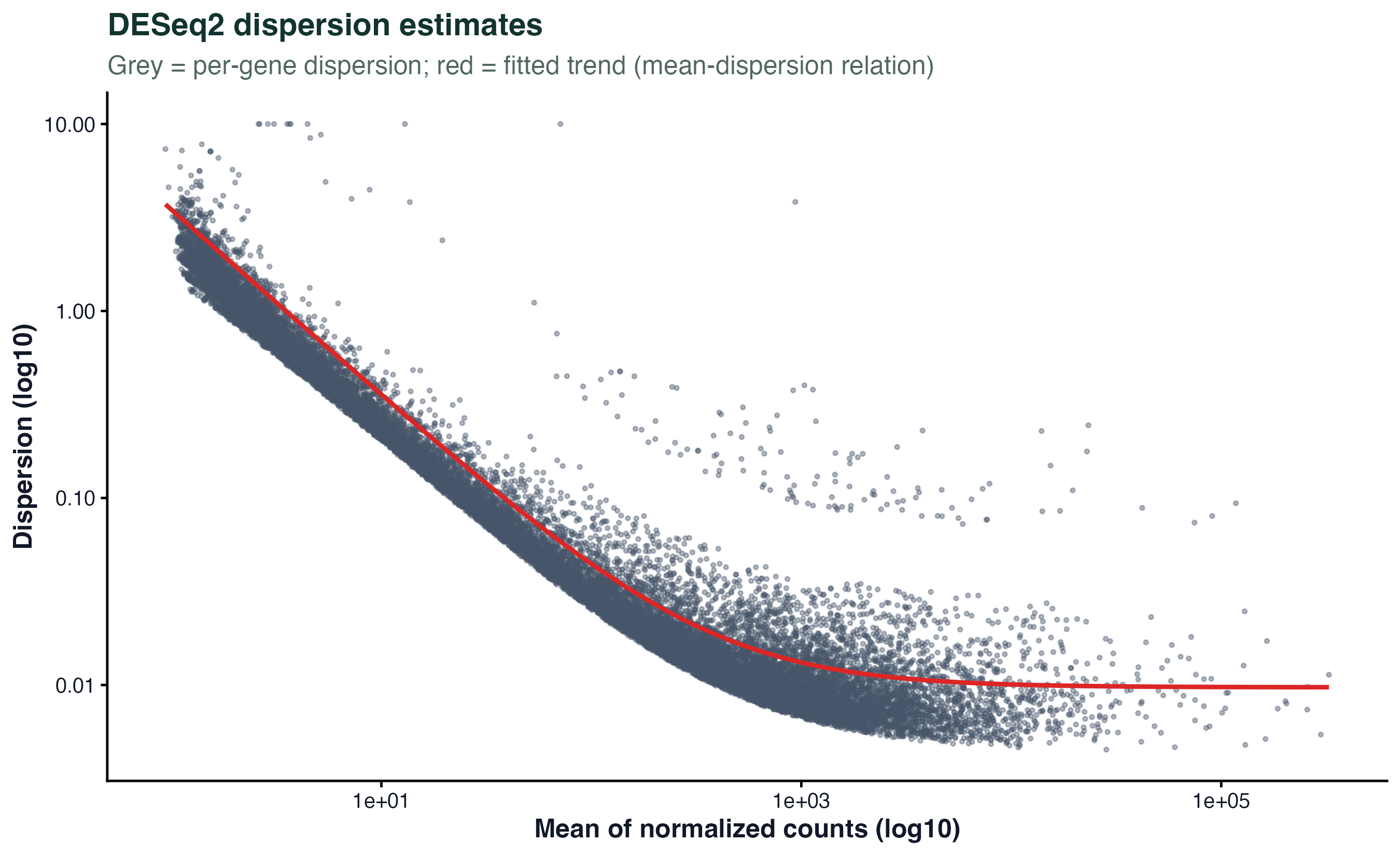
DESeq2 的核心是"用所有基因一起拟合 mean-dispersion 关系",低表达基因数据少、方差估计不稳,就借用趋势回到那条红色的拟合曲线上。灰色是每个基因的原始离散度,红线是拟合趋势。
看这张图主要是确认:趋势是不是随均值单调下降。如果红线走势很乱,可能说明样本里有严重的技术偏差。正常的曲线应该像这张一样,左下右上缓慢下降。
图 5:p 值分布与 MA 图
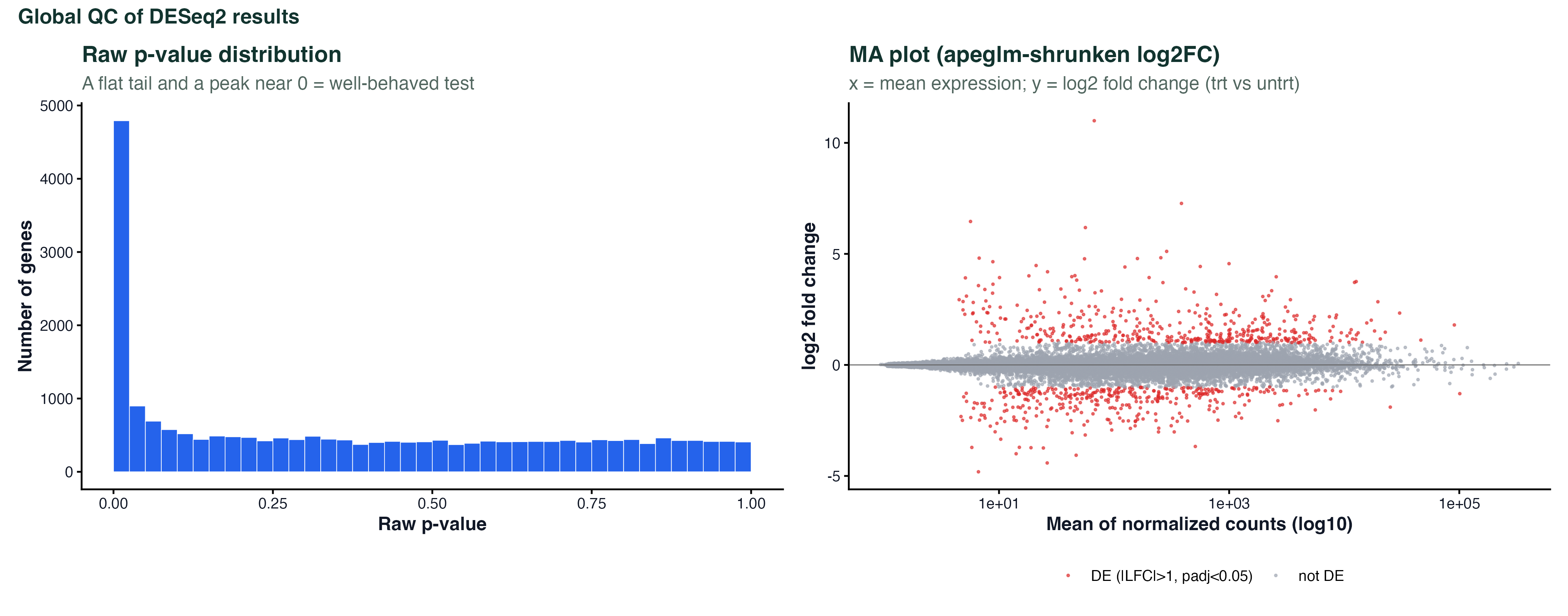
左边是原始 p 值直方图。一张健康的直方图长这样:00.05 一段突出(真正显著的基因),0.051 平坦分布(无效假设下的均匀分布)。如果直方图两端高中间低,说明离散度估计有问题;如果整体偏左(p 值都很小),可能是 design 漏了重要的协变量。
右边是 MA plot,横轴 log10(均值表达量),纵轴 log2 fold change。红点是显著 DE 基因(|LFC| > 1 且 padj < 0.05)。这里用的是 apeglm shrunken LFC,低表达基因(左侧)的 fold change 被往 0 拉,避免"表达量低 + 噪声大 → fold change 虚高"的假象。
图 6:Top DE 基因的归一化 counts
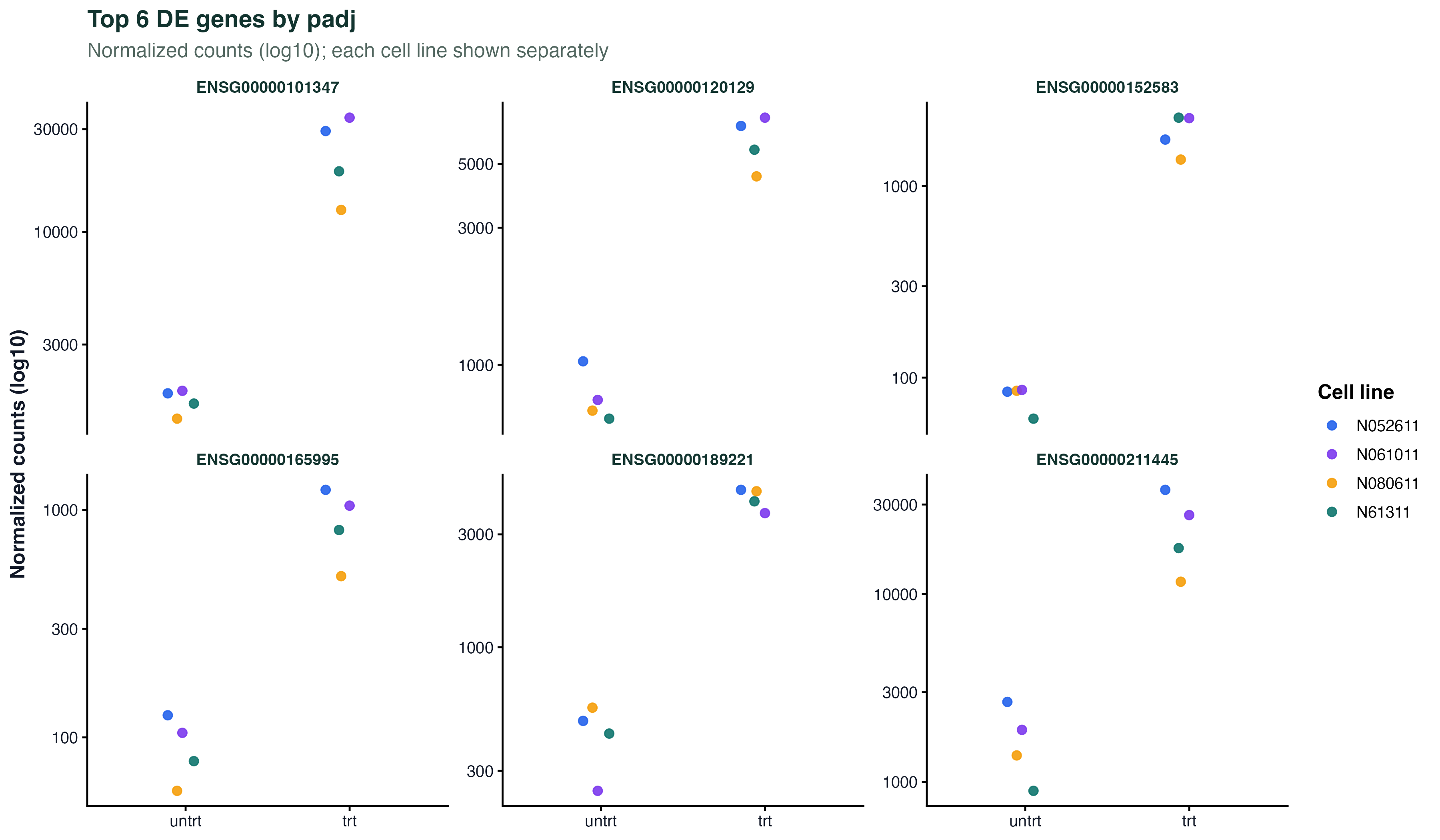
按 padj 排最前面的 6 个基因的 normalized counts,每条细胞系画成一个颜色。y 轴是 log10。每个基因都能看到 4 个 untrt 和 4 个 trt 点,处理组和对照组在 y 轴上分得很开,且不同细胞系的点虽然绝对值有差异,但处理效应方向一致 —— 这是 DESeq2 在控制了 cell 之后仍然能拿到强显著性的原因。
提取结果表
脚本末尾把 shrunken log2FC + 原始 padj 写成一张 TSV:
de_tbl <- as.data.frame(res_shrink) |>
tibble::rownames_to_column("gene_id") |>
mutate(padj_raw = results(dds, contrast = c("dex", "trt", "untrt"))$padj) |>
arrange(padj_raw)
write.table(de_tbl, "de_genes_airway.tsv", sep = "\t", quote = FALSE, row.names = FALSE)
这张表的列:gene_id、baseMean、log2FoldChange(shrunken)、lfcSE、pvalue、padj_raw。进 clusterProfiler 或 fgsea 做富集分析就用这张表。
套到自己数据上
脚本的前 15 行把 airway 换成自己的数据就行。常见两种输入:
# 输入 1:tximport 的结果
library(tximport)
txi <- tximport(files, type = "salmon", tx2gene = tx2gene)
dds <- DESeqDataSetFromTximport(txi, colData = samples, design = ~ batch + condition)
# 输入 2:featureCounts 的 counts 矩阵
count_mat <- read.delim("counts.tsv", row.names = 1)
samples <- read.delim("metadata/samples.tsv", row.names = 1)
dds <- DESeqDataSetFromMatrix(count_mat, colData = samples, design = ~ batch + condition)
其余的 DESeq() / results() / lfcShrink() 调用不用改。真实项目里最容易踩的坑:
- 样本表里
rownames要和 counts 矩阵的列名严格一致(包括顺序) - 如果样本有批次(不同测序 run、不同试剂盒批次),一定要加进 design
- 小于 3 对 3 的实验 DESeq2 能跑,但统计力很弱,不建议做复杂 design
常见坑
坑 1:把 TPM 当 counts 喂给 DESeq2
DESeqDataSetFromMatrix 检查不到这种错误 —— 因为 TPM 也是数值矩阵。但 dispersion 估计会完全失真,p 值可信度归零。Salmon / kallisto 输出的是 TPM 和 estimated counts,用 tximport(..., type = "salmon") 拿 counts 列,不要直接读 quant.sf 的 TPM。
坑 2:design 公式里把要检验的变量放前面
design = ~ dex + cell 和 design = ~ cell + dex 的拟合系数完全一样,但 results() �默认提取最后一个变量的对比。要检验的变量放最后是社区惯例,否则 results() 会拿错对比。
坑 3:用未 relevel 的因子默认顺序
R 里因子默认按字母顺序排("trt" 在 "untrt" 前面),这导致 DESeq2 把 "trt" 当参考、"untrt" 当对比 —— log2FC 符号正好反过来。养成习惯:建立 dds 之前显式 relevel(factor, ref = "untrt"),或在 results() 里 contrast = c("dex","trt","untrt") 写清楚。
坑 4:用原始 log2FC 做下游而不是 shrunken
低表达基因(baseMean < 10)的 LFC 噪声很大,会出现 |LFC| > 5 但 padj 接近 1 的情况。做下游分析(GSEA 排序、可视化、报告)一律用 lfcShrink(..., type = "apeglm"),原始 LFC 只在写 supp table 时保留。
坑 5:直接信 padj < 0.05,不看 p 值分布
p 值直方图是一个免费的"模型质量检查":
- 健康:左侧高峰 + 右侧均匀
- U 型:模型欠拟合(design 漏了变量)或数据有奇异样本
- 整体偏左:有未控制的混杂因子
跑完 DESeq2 务必 hist(res$pvalue) 一秒钟看一眼,比单看 padj 信息量大得多。
下载资源
不想本地装环境?在 BioF3 上跑
下一步
接着深入:
- 03 功能富集:GO / KEGG / GSEA — 拿到差异基因表后第一件事是富集分析,把基因翻译成通路
- 04 火山图、热图与富集可视化 — 同一份 DE 结果可以做出能直接发表的 6 张图
横向延伸:
- 05 多时间点与 LRT — 你的设计里有时间序列 / 剂量响应时,Wald 检验不够,要换 LRT
- 06 批次效应 — 多批次数据的 design 公式怎么写
- 07 多工具对比 — 想知道 edgeR / limma-voom 的结果会不会很不一样
参考资源
离线资料下载
手册 HTML / PDF 已在后台预生成,点击后直接下载网站静态资源。