模块04:多样本数据整合
本模块将介绍如何整合来自不同批次、不同条件或不同实验的单细胞数据。
学习目标
- 理解批次效应的来源和影响
- 掌握多种数据整合方法
- 学会评估整合效果
- 进行整合后的下游分析
为什么需要数据整合?
AI 时代的学习方式
Vibe coding 带来极大的便利,能否用好工具需要思想的指引。如果想复现这些分析,建议下载完整脚本学习。
(15 KB)
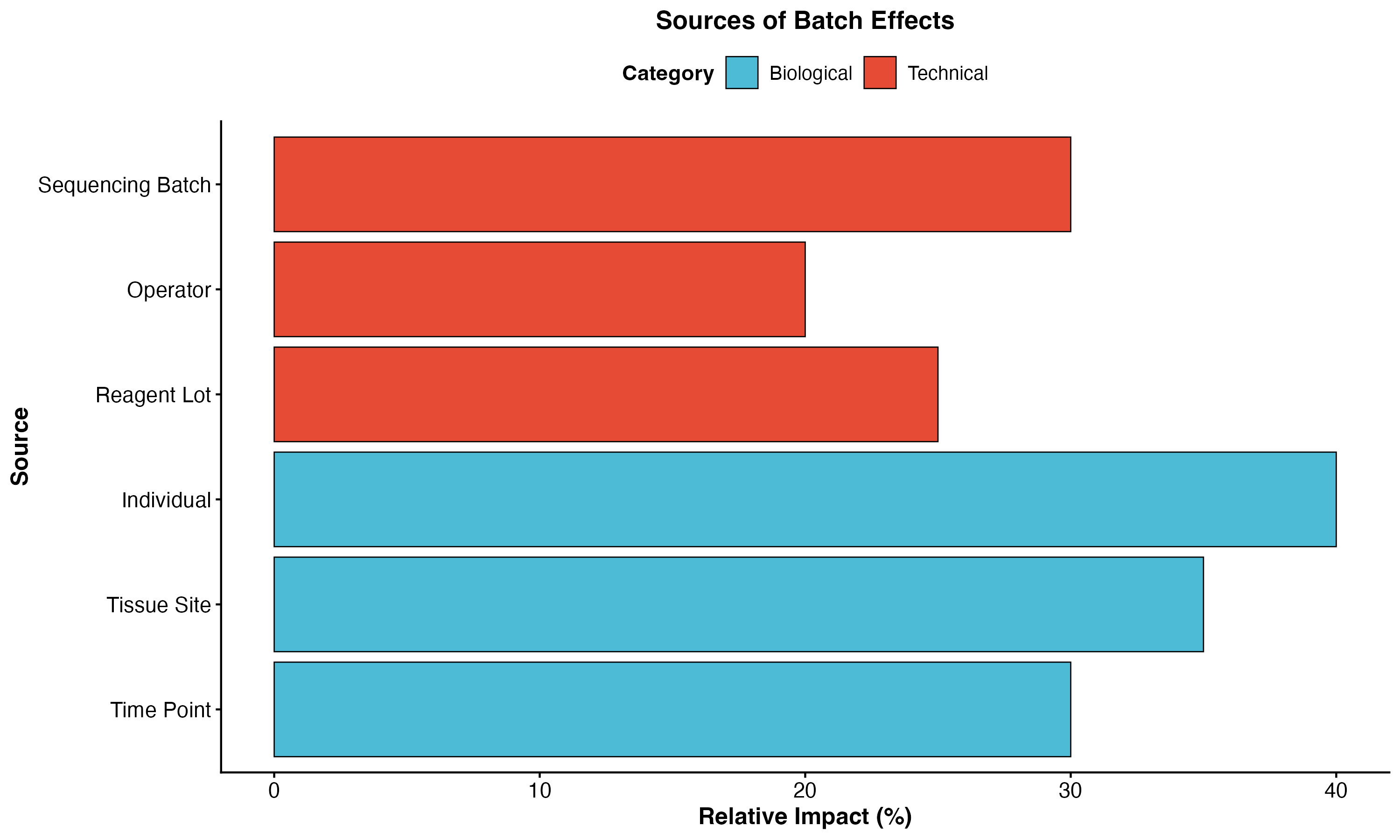
图 1:批次效应的来源。展示了技术批次和生物学批次的不同来源及其相对影响。
批次效应的来源
-
技术批次
- 不同测序批次
- 不同实验人员
- 不同试剂批次
-
生物学批次
- 不同个体
- 不同组织部位
- 不同时间点
批次效应的影响
- 掩盖真实的生物学差异
- 产生虚假的细胞亚群
- 影响差异表达分析
整合方法概览
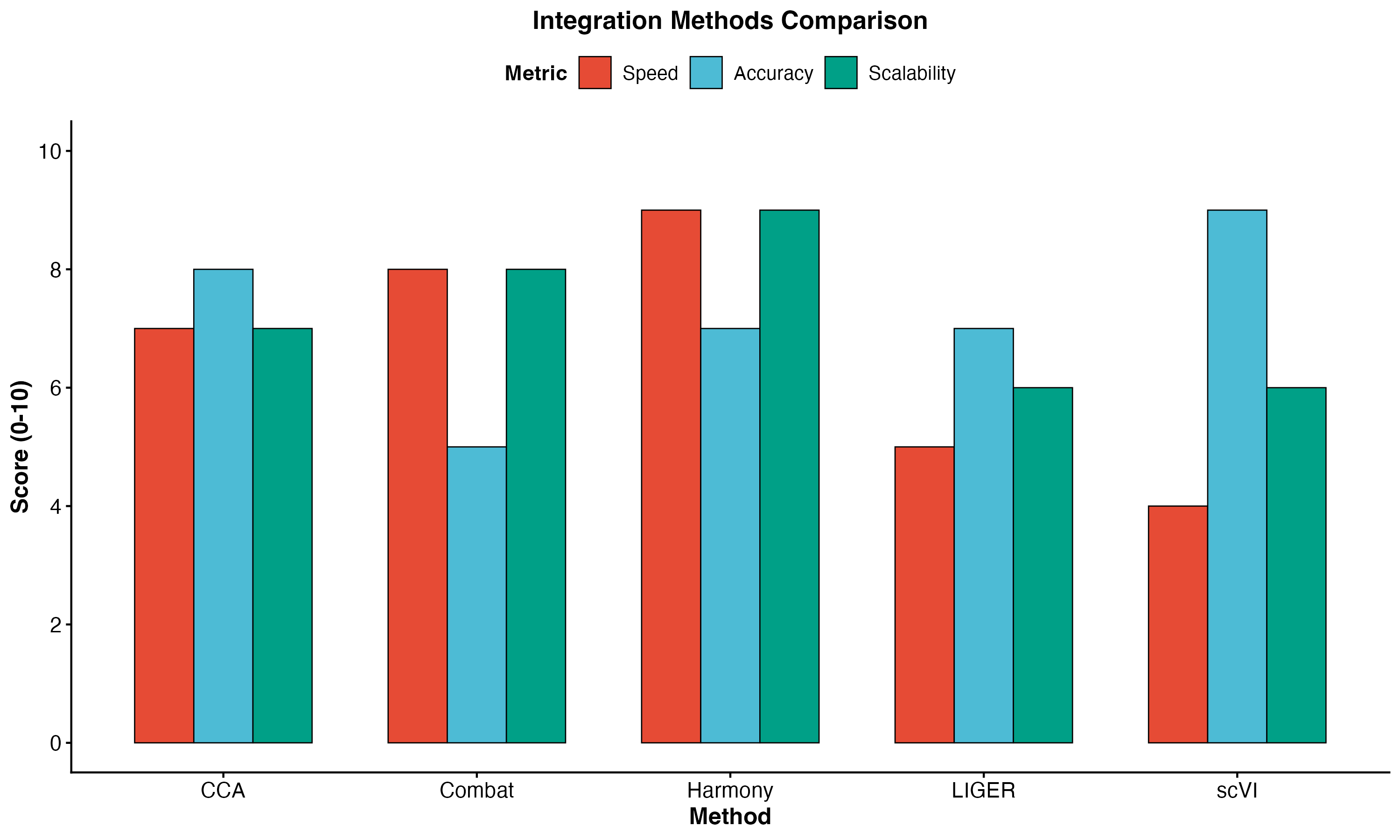
图 2:不同整合方法的对比。展示了 CCA、Harmony、scVI、LIGER 和 Combat 五种方法在速度、准确性和可扩展性三个维度的评分。
| 方法 | 工具 | 优点 | 缺点 |
|---|---|---|---|
| CCA | Seurat | 快速,效果好 | 需要共享细胞类型 |
| Harmony | Harmony | 非常快速 | 可能过度校正 |
| scVI | scvi-tools | 深度学习,强大 | 计算密集 |
| LIGER | LIGER | 适合跨物种 | 较慢 |
| Combat | sva | 简单 | 效果一般 |
使用 Seurat 整合
准备数据
library(Seurat)
# 读取多个样本
sample1 <- Read10X("sample1/filtered_feature_bc_matrix/")
sample2 <- Read10X("sample2/filtered_feature_bc_matrix/")
sample3 <- Read10X("sample3/filtered_feature_bc_matrix/")
# 创建 Seurat 对象
pbmc1 <- CreateSeuratObject(sample1, project = "sample1")
pbmc2 <- CreateSeuratObject(sample2, project = "sample2")
pbmc3 <- CreateSeuratObject(sample3, project = "sample3")
# 合并到列表
pbmc.list <- list(pbmc1, pbmc2, pbmc3)
标准流程(每个样本)
# 对每个样本进行标准化和特征选择
pbmc.list <- lapply(X = pbmc.list, FUN = function(x) {
x <- NormalizeData(x)
x <- FindVariableFeatures(x, selection.method = "vst", nfeatures = 2000)
})
方法1:使用 CCA 整合
# 选择整合特征
features <- SelectIntegrationFeatures(object.list = pbmc.list)
# 寻找整合锚点
pbmc.anchors <- FindIntegrationAnchors(
object.list = pbmc.list,
anchor.features = features
)
# 整合数据
pbmc.combined <- IntegrateData(anchorset = pbmc.anchors)
# 切换到整合后的数据
DefaultAssay(pbmc.combined) <- "integrated"
# 标准流程
pbmc.combined <- ScaleData(pbmc.combined, verbose = FALSE)
pbmc.combined <- RunPCA(pbmc.combined, npcs = 30, verbose = FALSE)
pbmc.combined <- RunUMAP(pbmc.combined, reduction = "pca", dims = 1:30)
pbmc.combined <- FindNeighbors(pbmc.combined, reduction = "pca", dims = 1:30)
pbmc.combined <- FindClusters(pbmc.combined, resolution = 0.5)
# 可视化
DimPlot(pbmc.combined, reduction = "umap", group.by = "orig.ident")
DimPlot(pbmc.combined, reduction = "umap", label = TRUE)
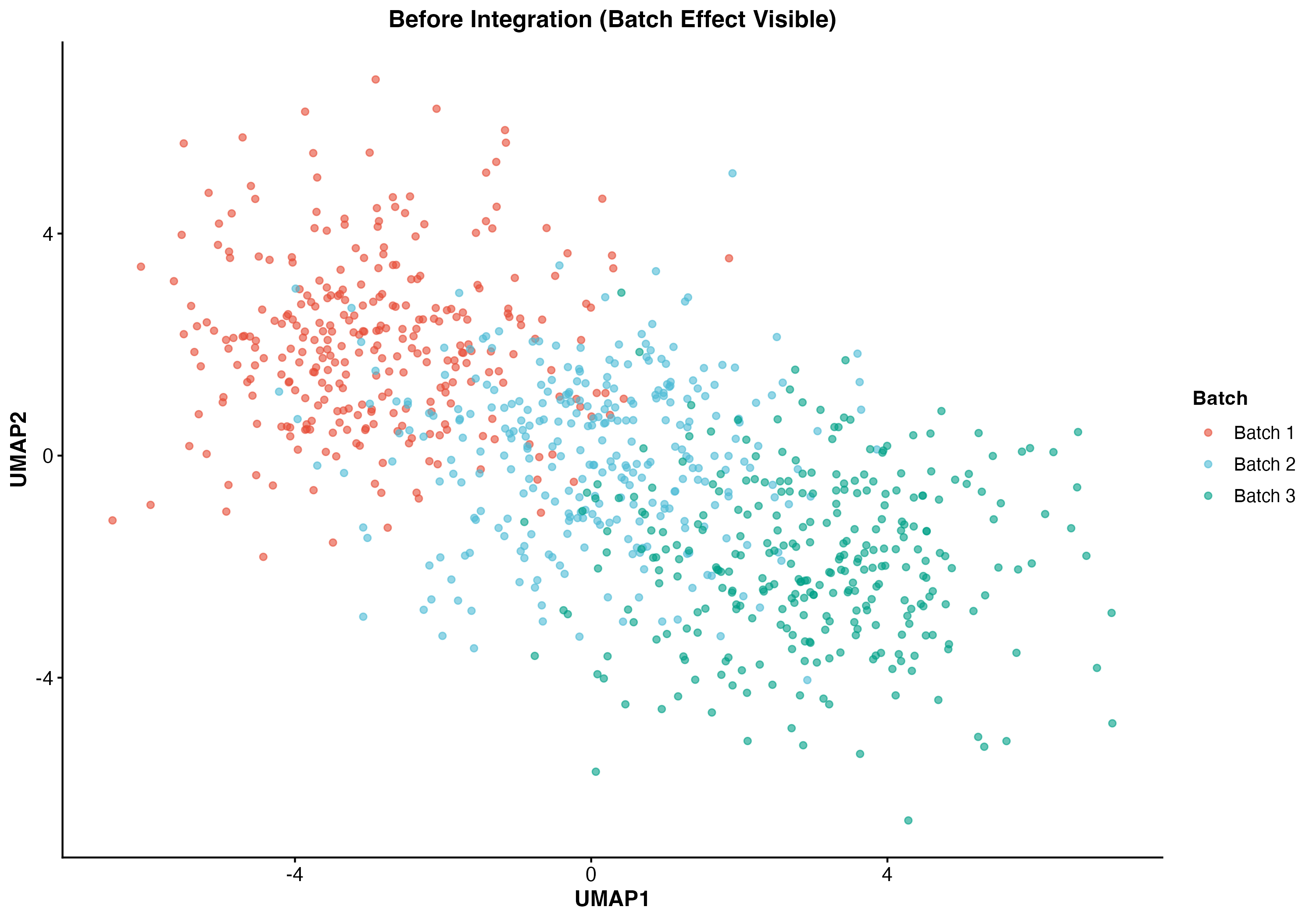
图 3:整合前的 UMAP 图。展示了明显的批次效应,不同批次的细胞分离成不同的簇。
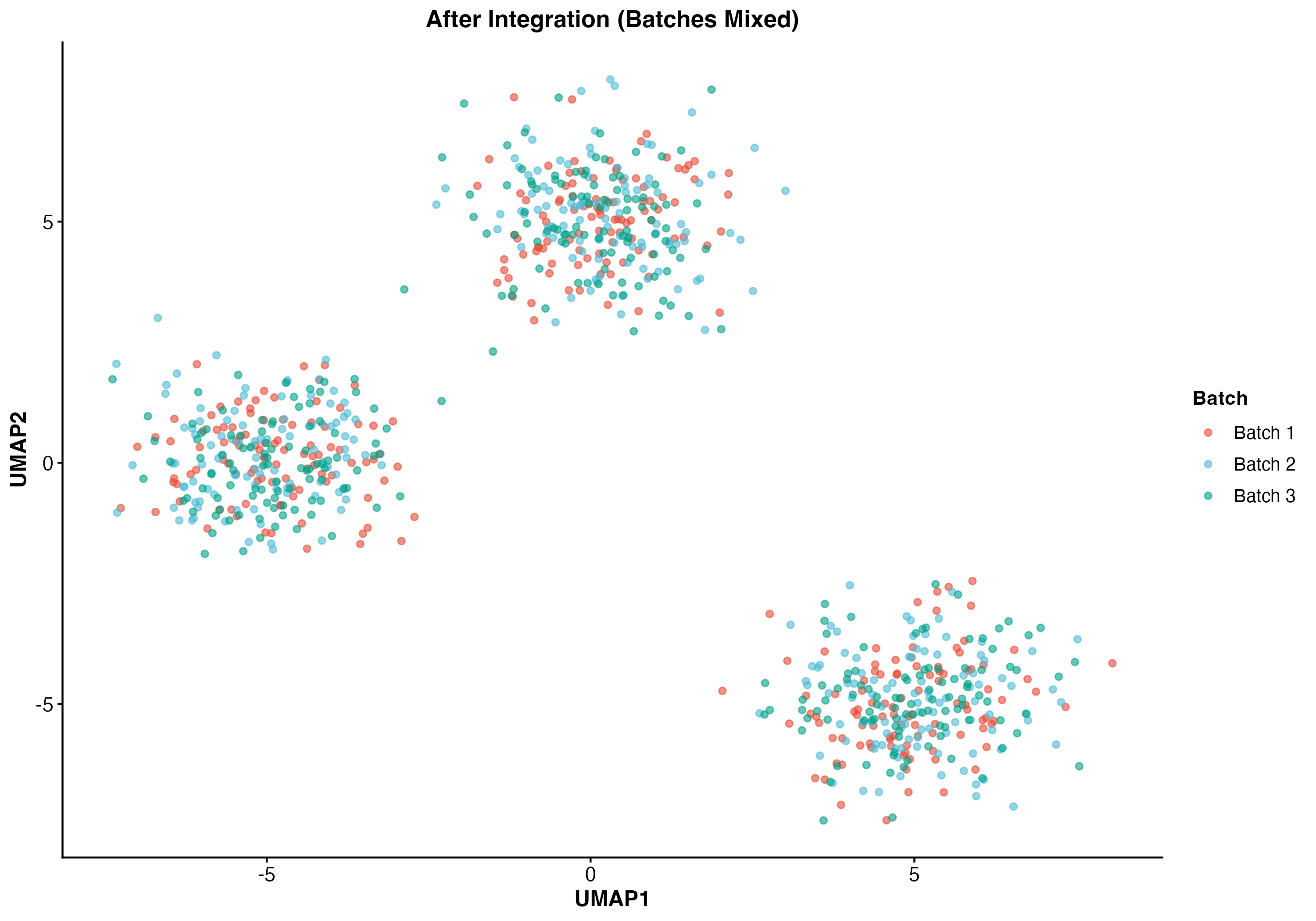
图 4:整合后的 UMAP 图。批次效应被消除,不同批次的细胞混合在一起。
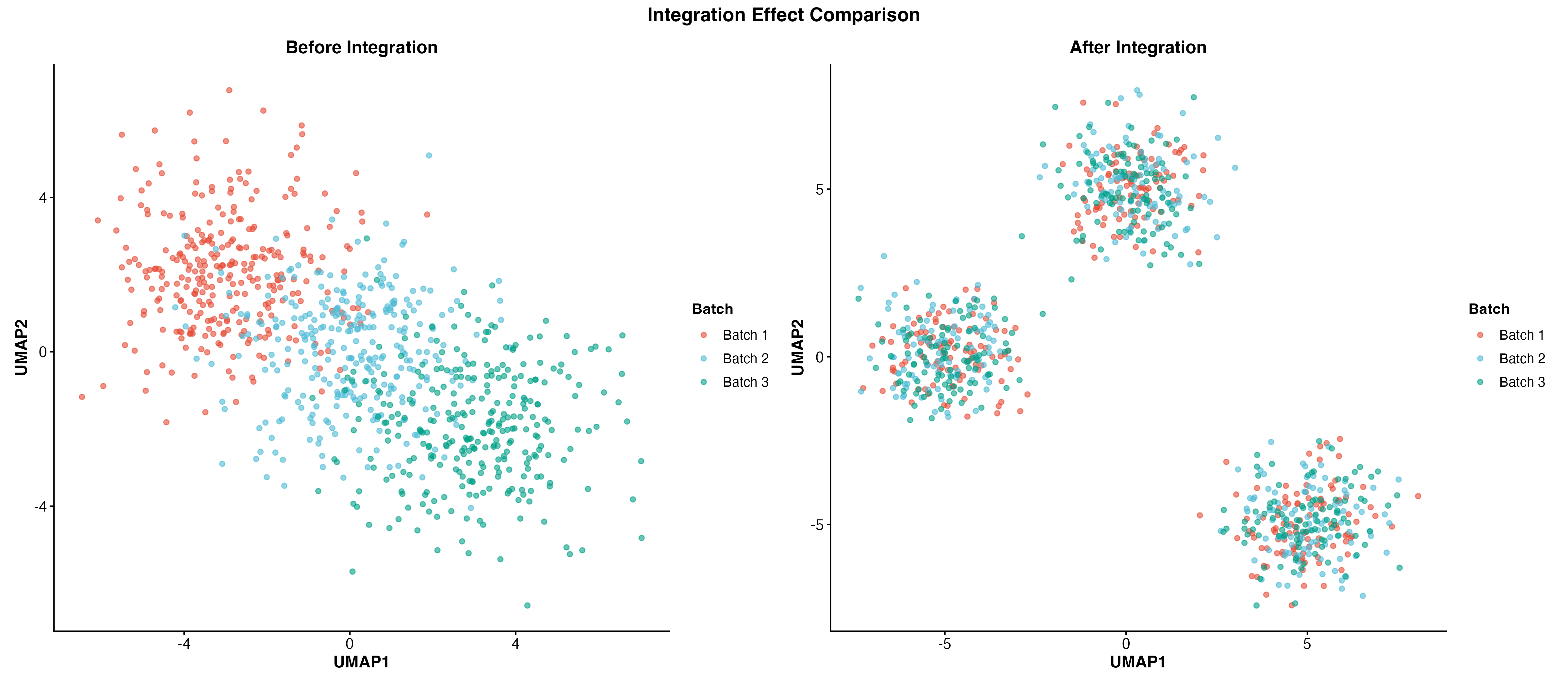
图 5:整合前后对比。左图显示整合前的批次分离,右图显示整合后的批次混合。
方法2:使用 SCTransform 整合
# 对每个样本运行 SCTransform
pbmc.list <- lapply(X = pbmc.list, FUN = SCTransform)
# 选择整合特征
features <- SelectIntegrationFeatures(object.list = pbmc.list, nfeatures = 3000)
# 准备整合
pbmc.list <- PrepSCTIntegration(object.list = pbmc.list, anchor.features = features)
# 寻找锚点
pbmc.anchors <- FindIntegrationAnchors(
object.list = pbmc.list,
normalization.method = "SCT",
anchor.features = features
)
# 整合
pbmc.combined.sct <- IntegrateData(anchorset = pbmc.anchors, normalization.method = "SCT")
# 下游分析
pbmc.combined.sct <- RunPCA(pbmc.combined.sct, verbose = FALSE)
pbmc.combined.sct <- RunUMAP(pbmc.combined.sct, reduction = "pca", dims = 1:30)
使用 Harmony 整合
library(harmony)
# 合并所有样本(不整合)
pbmc.merged <- merge(pbmc1, y = c(pbmc2, pbmc3),
add.cell.ids = c("S1", "S2", "S3"))
# 标准流程
pbmc.merged <- NormalizeData(pbmc.merged)
pbmc.merged <- FindVariableFeatures(pbmc.merged)
pbmc.merged <- ScaleData(pbmc.merged)
pbmc.merged <- RunPCA(pbmc.merged)
# 运行 Harmony
pbmc.harmony <- RunHarmony(pbmc.merged, group.by.vars = "orig.ident")
# 使用 Harmony 降维结果
pbmc.harmony <- RunUMAP(pbmc.harmony, reduction = "harmony", dims = 1:30)
pbmc.harmony <- FindNeighbors(pbmc.harmony, reduction = "harmony", dims = 1:30)
pbmc.harmony <- FindClusters(pbmc.harmony, resolution = 0.5)
# 可视化
DimPlot(pbmc.harmony, reduction = "umap", group.by = "orig.ident")
DimPlot(pbmc.harmony, reduction = "umap", label = TRUE)
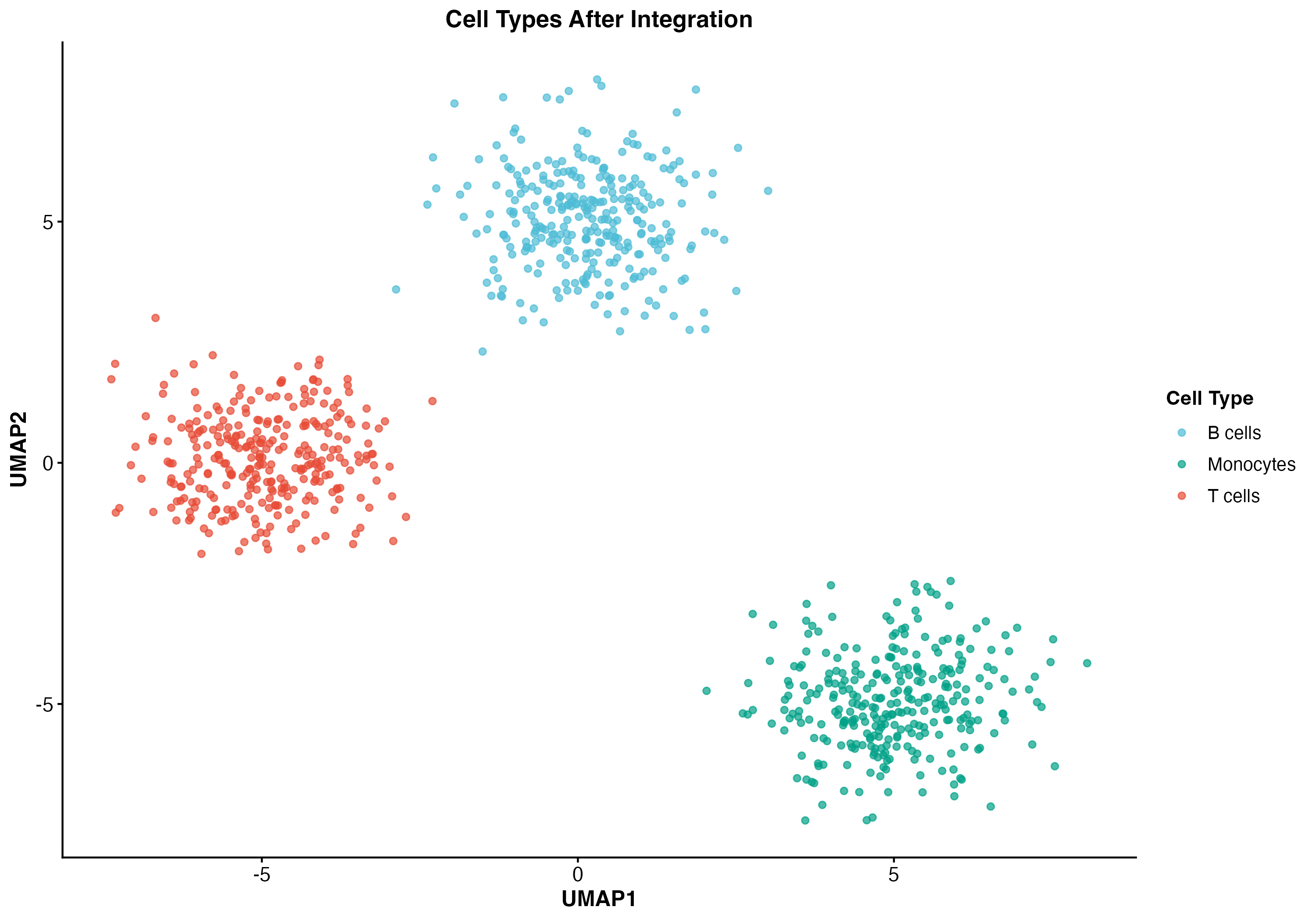
图 6:整合后的细胞类型分布。展示了 T 细胞、B 细胞和单核细胞在 UMAP 空间中的分布。
使用 Scanpy 整合
使用 scVI
import scanpy as sc
import scvi
# 读取数据
adata1 = sc.read_10x_mtx('sample1/')
adata2 = sc.read_10x_mtx('sample2/')
adata3 = sc.read_10x_mtx('sample3/')
# 添加批次信息
adata1.obs['batch'] = 'sample1'
adata2.obs['batch'] = 'sample2'
adata3.obs['batch'] = 'sample3'
# 合并
adata = adata1.concatenate(adata2, adata3)
# 预处理
sc.pp.filter_cells(adata, min_genes=200)
sc.pp.filter_genes(adata, min_cells=3)
sc.pp.normalize_total(adata, target_sum=1e4)
sc.pp.log1p(adata)
sc.pp.highly_variable_genes(adata, n_top_genes=2000, batch_key='batch')
# 设置 scVI
scvi.model.SCVI.setup_anndata(adata, batch_key='batch')
# 训练模型
model = scvi.model.SCVI(adata)
model.train()
# 获取整合后的表示
adata.obsm['X_scVI'] = model.get_latent_representation()
# 下游分析
sc.pp.neighbors(adata, use_rep='X_scVI')
sc.tl.umap(adata)
sc.tl.leiden(adata)
# 可视化
sc.pl.umap(adata, color=['batch', 'leiden'])
使用 Harmony (Python)
import scanpy as sc
import scanpy.external as sce
# 合并数据
adata = adata1.concatenate(adata2, adata3)
# 预处理
sc.pp.normalize_total(adata)
sc.pp.log1p(adata)
sc.pp.highly_variable_genes(adata)
sc.pp.scale(adata)
sc.tl.pca(adata)
# 运行 Harmony
sce.pp.harmony_integrate(adata, 'batch')
# 下游分析
sc.pp.neighbors(adata, use_rep='X_pca_harmony')
sc.tl.umap(adata)
sc.tl.leiden(adata)
# 可视化
sc.pl.umap(adata, color=['batch', 'leiden'])
评估整合效果
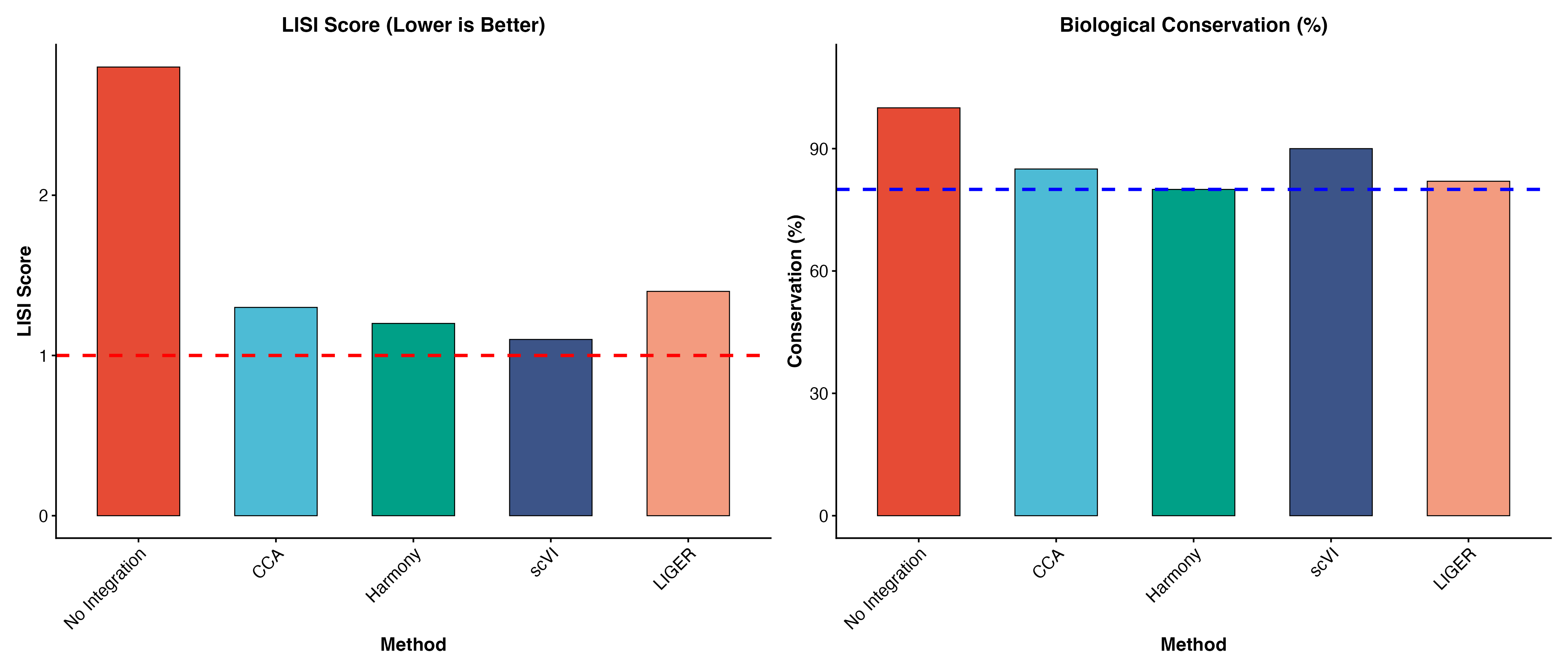
图 7:整合质量评估指标。左图显示 LISI 分数(越接近 1 越好),右图显示生物学信息保留率。
1. 可视化评估
# 整合前后对比
p1 <- DimPlot(pbmc.merged, reduction = "umap", group.by = "orig.ident") +
ggtitle("Before Integration")
p2 <- DimPlot(pbmc.combined, reduction = "umap", group.by = "orig.ident") +
ggtitle("After Integration")
p1 + p2
2. 定量评估
library(lisi)
# 计算 LISI 分数
# 值越接近1,整合越好
lisi_scores <- compute_lisi(
pbmc.combined@reductions$pca@cell.embeddings,
pbmc.combined@meta.data,
c("orig.ident")
)
3. 保留生物学差异
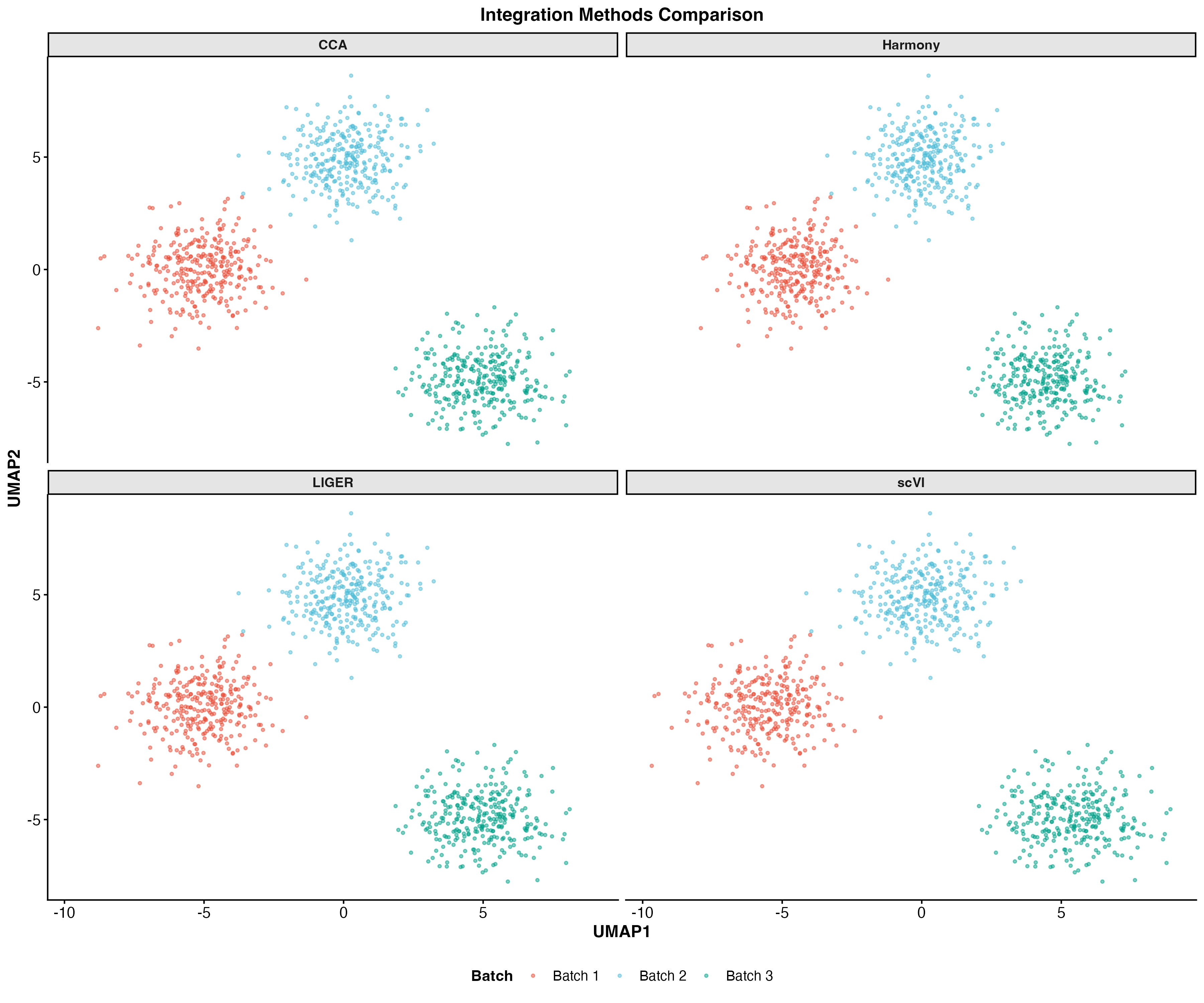
图 8:不同整合方法的 UMAP 对比。展示了 CCA、Harmony、scVI 和 LIGER 四种方法的整合效果。
# 检查已知的细胞类型标志基因
FeaturePlot(pbmc.combined, features = c("CD3D", "CD14", "MS4A1"))
# 确保不同条件的差异仍然存在
DimPlot(pbmc.combined, split.by = "condition")
图 9:基因表达分布的保留。展示了整合前后基因表达在不同批次间的分布变化。
整合后的差异分析
比较不同条件
# 切换回原始数据进行差异分析
DefaultAssay(pbmc.combined) <- "RNA"
# 比较不同条件下的同一细胞类型
Idents(pbmc.combined) <- "cell_type"
condition_markers <- FindMarkers(
pbmc.combined,
ident.1 = "CD4 T",
group.by = "condition",
subset.ident = "treated",
test.use = "MAST"
)
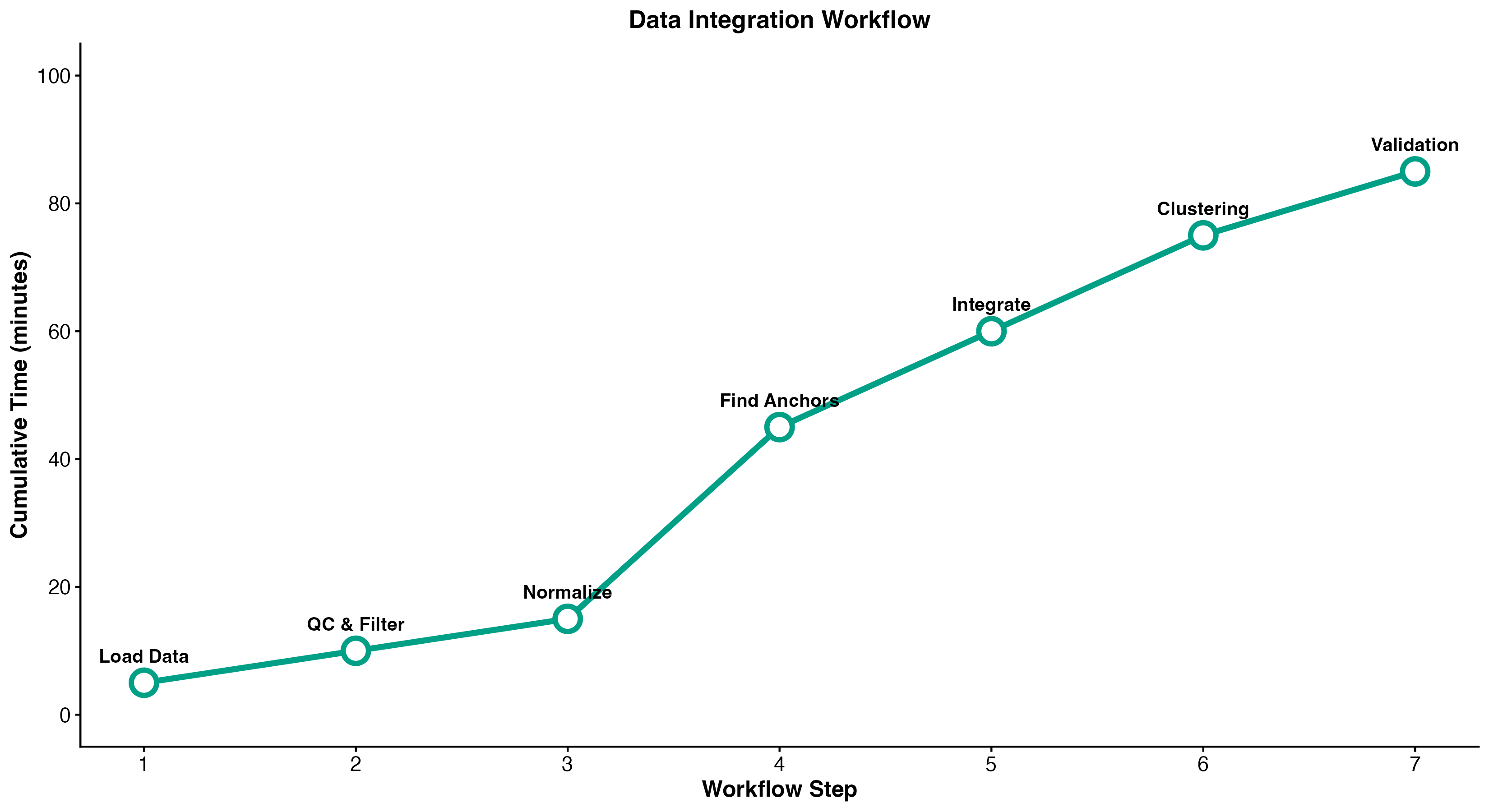
图 10:数据整合完整工作流程。展示了从数据加载到验证的 7 个主要步骤及累计时间。
最佳实践
-
选择合适的整合方法
- 样本数少:CCA
- 样本数多:Harmony
- 需要最佳效果:scVI
-
保留原始数据
- 整合用于聚类和可视化
- 差异分析使用原始数据
-
验证整合效果
- 批次效应是否消除
- 生物学差异是否保留
-
谨慎解释结果
- 过度整合可能掩盖真实差异
- 结合生物学知识判断