模块03:质量控制、聚类与细胞类型注释
本模块将介绍单细胞数据分析的核心步骤:质量控制、数据标准化、降维、聚类和细胞类型注释。
学习目标
完成本模块后,你将能够:
- 进行严格的质量控制,过滤低质量细胞
- 标准化和归一化单细胞数据
- 使用 PCA 和 UMAP 进行降维
- 进行细胞聚类分析
- 注释细胞类型
- 识别细胞类型标志基因
- 进行功能富集分析
前置知识
- 完成模块02(Cell Ranger 数据处理)
- R 或 Python 编程基础
- 基本的统计学知识
推荐实践数据
本模块建议使用 PBMC 3k 真实数据进行练习。该数据体积小、下载快,适合完整跑通质量控制、标准化、降维、聚类和细胞类型注释流程。
分析流程概览
图 1:单细胞分析完整流程。展示了从原始数据到细胞类型注释的 8 个主要步骤及细胞保留率。
原始表达矩阵
↓
质量控制 (QC)
↓
数据标准化
↓
特征选择(高变异基因)
↓
降维 (PCA)
↓
聚类分析
↓
非线性降维 (UMAP/t-SNE)
↓
细胞类型注释
↓
差异表达分析
↓
功能富集分析
环境准备
R 环境
# 安装必需的包
install.packages("Seurat")
install.packages("dplyr")
install.packages("ggplot2")
# Bioconductor 包
if (!require("BiocManager", quietly = TRUE))
install.packages("BiocManager")
BiocManager::install("SingleR")
BiocManager::install("celldex")
BiocManager::install("clusterProfiler")
# 加载包
library(Seurat)
library(dplyr)
library(ggplot2)
Python 环境
# 安装必需的包
pip install scanpy python-igraph leidenalg
# 导入包
import scanpy as sc
import pandas as pd
import numpy as np
import matplotlib.pyplot as plt
# 设置
sc.settings.verbosity = 3
sc.settings.set_figure_params(dpi=80, facecolor='white')
数据加载
使用 Seurat (R)
library(Seurat)
# 读取 10x 数据
data_dir <- "path/to/filtered_feature_bc_matrix/"
data <- Read10X(data.dir = data_dir)
# 创建 Seurat 对象
pbmc <- CreateSeuratObject(
counts = data,
project = "PBMC3k",
min.cells = 3, # 基因至少在3个细胞中表达
min.features = 200 # 细胞至少表达200个基因
)
# 查看对象
pbmc
使用 Scanpy (Python)
import scanpy as sc
# 读取 10x 数据
adata = sc.read_10x_mtx(
'path/to/filtered_feature_bc_matrix/',
var_names='gene_symbols',
cache=True
)
# 查看对象
print(adata)
质量控制 (QC)
质量指标
1. 每个细胞的基因数 (nFeature/n_genes)
含义: 细胞中检测到的基因数量
正常范围:
- 200 - 6,000 基因
异常情况:
- 过低 (< 200): 空液滴、破损细胞
- 过高 (> 6,000): 双细胞
2. 每个细胞的 UMI 数 (nCount/n_counts)
含义: 细胞中检测到的总 UMI 数
正常范围:
- 500 - 50,000 UMI
异常情况:
- 过低: 测序深度不足
- 过高: 双细胞
3. 线粒体基因比例 (percent.mt)
含义: 线粒体基因表达占总表达的比例
正常范围:
- < 5-10%
异常情况:
- 过高: 细胞死亡、细胞质流失
计算 QC 指标
Seurat (R)
# 计算线粒体基因比例
pbmc[["percent.mt"]] <- PercentageFeatureSet(pbmc, pattern = "^MT-")
# 计算核糖体基因比例(可选)
pbmc[["percent.rb"]] <- PercentageFeatureSet(pbmc, pattern = "^RP[SL]")
# 查看 QC 指标
head(pbmc@meta.data)
Scanpy (Python)
# 计算线粒体基因比例
adata.var['mt'] = adata.var_names.str.startswith('MT-')
sc.pp.calculate_qc_metrics(
adata,
qc_vars=['mt'],
percent_top=None,
log1p=False,
inplace=True
)
# 查看 QC 指标
adata.obs.head()
可视化 QC 指标
Vibe coding 带来极大的便利,能否用好工具需要思想的指引。如果想复现这些分析,建议下载完整脚本学习。
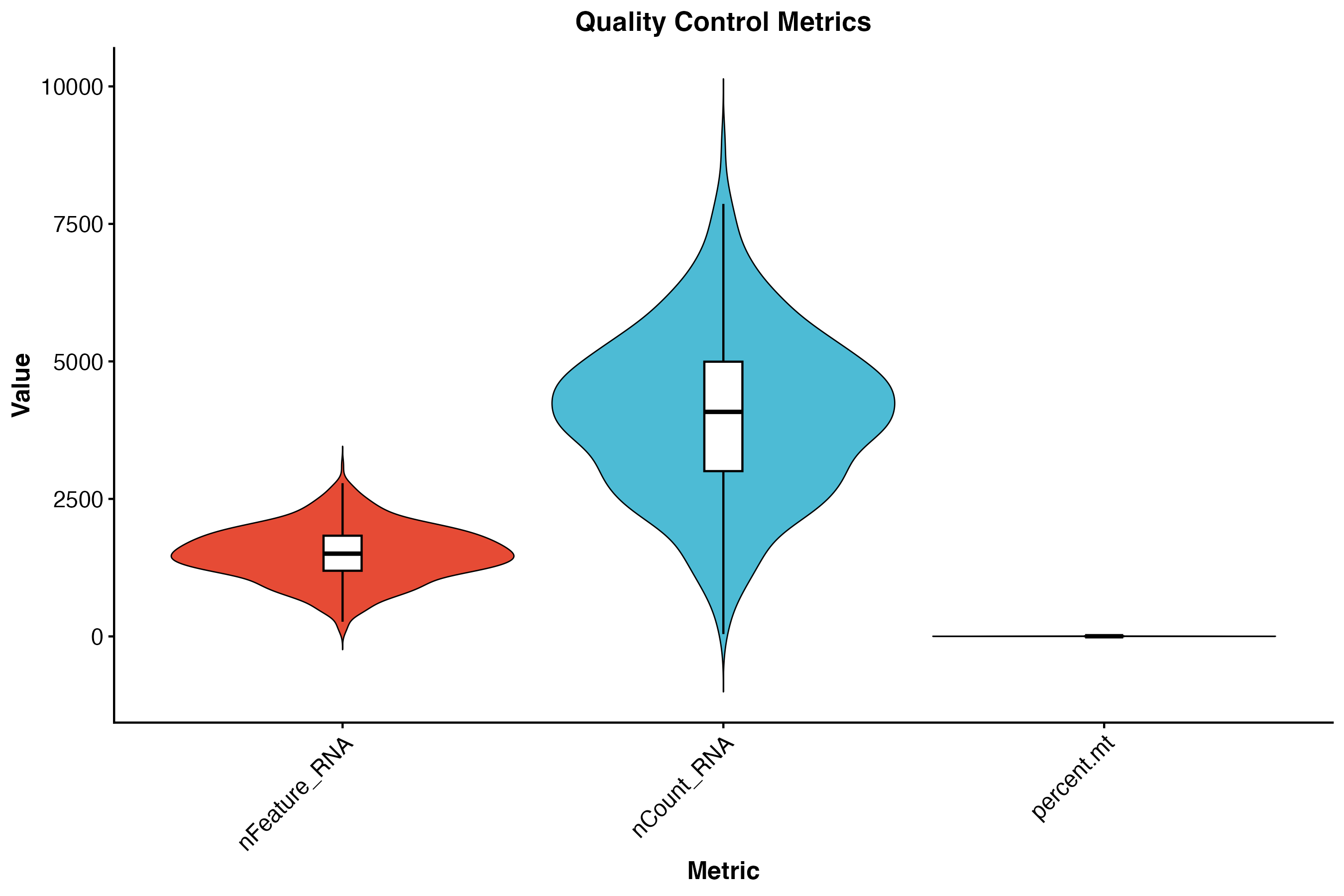
图 2:质量控制指标小提琴图。展示了基因数、UMI 数和线粒体基因比例的分布情况。
Seurat (R)
# 小提琴图
VlnPlot(pbmc,
features = c("nFeature_RNA", "nCount_RNA", "percent.mt"),
ncol = 3)
# 散点图
plot1 <- FeatureScatter(pbmc, feature1 = "nCount_RNA", feature2 = "percent.mt")
plot2 <- FeatureScatter(pbmc, feature1 = "nCount_RNA", feature2 = "nFeature_RNA")
plot1 + plot2
Scanpy (Python)
# 小提琴图
sc.pl.violin(adata,
['n_genes_by_counts', 'total_counts', 'pct_counts_mt'],
jitter=0.4,
multi_panel=True)
# 散点图
sc.pl.scatter(adata, x='total_counts', y='pct_counts_mt')
sc.pl.scatter(adata, x='total_counts', y='n_genes_by_counts')
过滤细胞
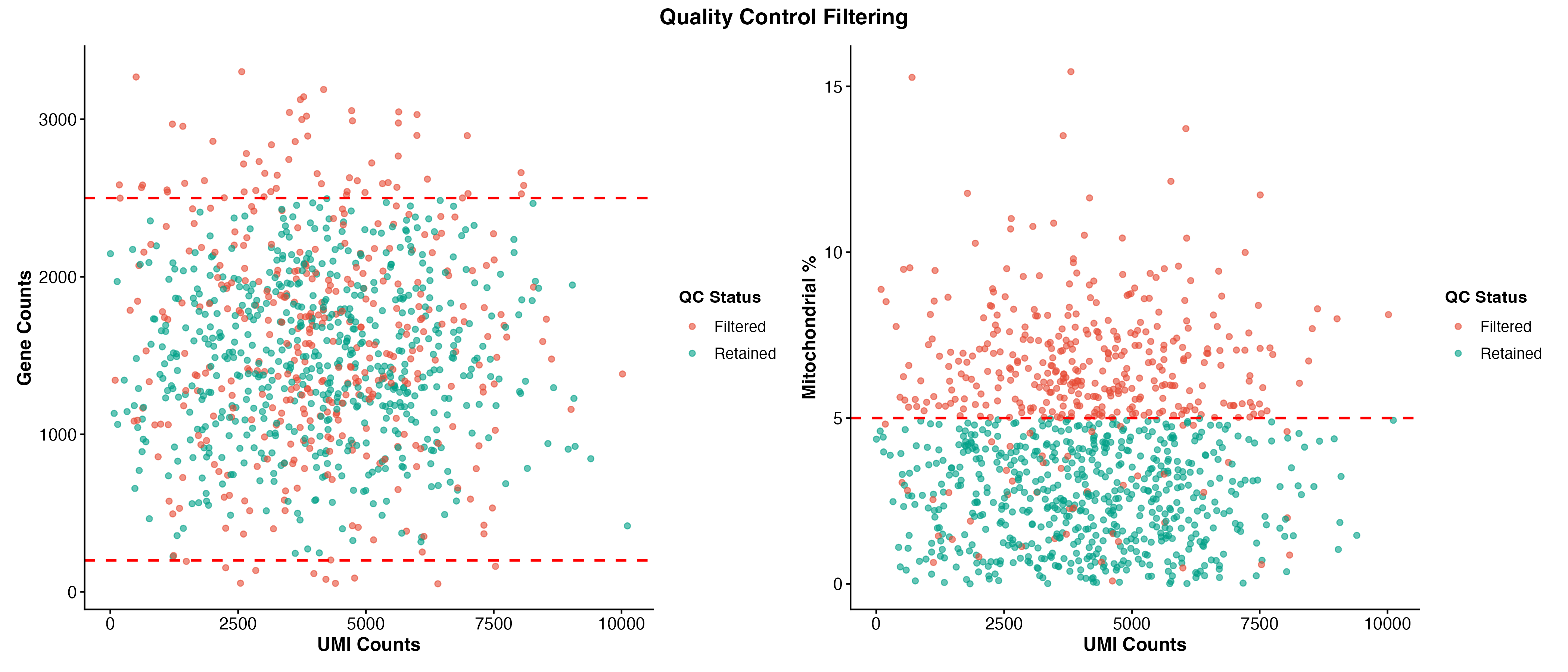
图 3:质量控制过滤散点图。左图展示 UMI 数与基因数的关系,右图展示 UMI 数与线粒体基因比例的关系。红色虚线表示过滤阈值,绿色点为保留的细胞,红色点为过滤的细胞。
Seurat (R)
# 设置过滤阈值
pbmc <- subset(pbmc, subset =
nFeature_RNA > 200 &
nFeature_RNA < 2500 &
percent.mt < 5
)
# 查看过滤后的细胞数
pbmc
Scanpy (Python)
# 过滤细胞
sc.pp.filter_cells(adata, min_genes=200)
sc.pp.filter_cells(adata, max_genes=2500)
# 过滤线粒体基因比例高的细胞
adata = adata[adata.obs.pct_counts_mt < 5, :]
# 过滤基因(至少在3个细胞中表达)
sc.pp.filter_genes(adata, min_cells=3)
# 查看过滤后的数据
print(adata)
数据标准化
为什么需要标准化?
- 消除测序深度差异
- 使不同细胞之间可比较
- 稳定方差
Seurat 标准化
# LogNormalize 方法(默认)
pbmc <- NormalizeData(pbmc,
normalization.method = "LogNormalize",
scale.factor = 10000)
# 或使用 SCTransform(推荐)
pbmc <- SCTransform(pbmc,
vars.to.regress = "percent.mt",
verbose = FALSE)
Scanpy 标准化
# 标准化到每个细胞总计数为 10,000
sc.pp.normalize_total(adata, target_sum=1e4)
# Log 转换
sc.pp.log1p(adata)
# 保存原始数据
adata.raw = adata
特征选择(高变异基因)
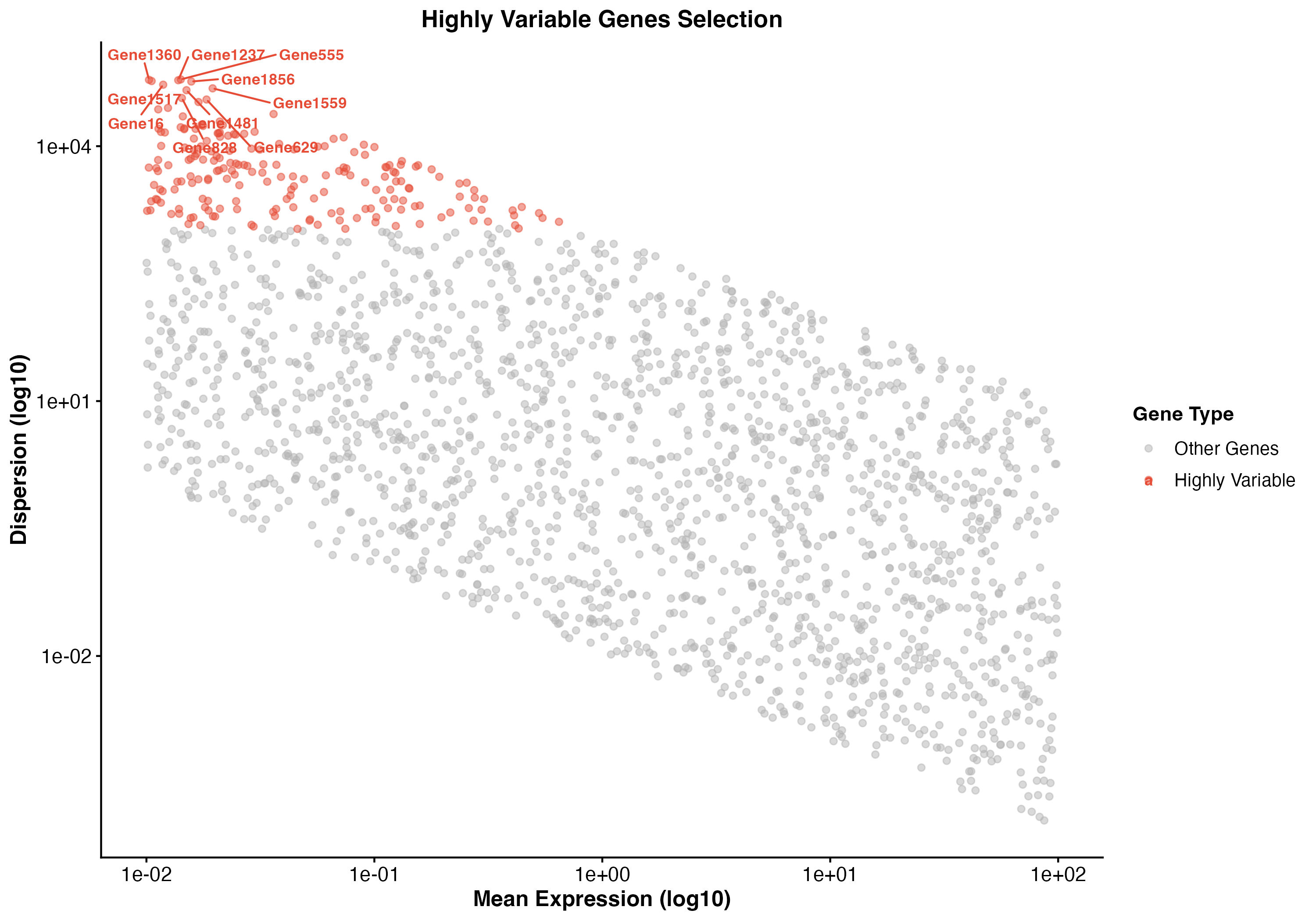
图 4:高变异基因选择。展示了基因的平均表达量与离散度的关系,红色点为高变异基因,标注了前 10 个高变异基因。
为什么选择高变异基因?
- 减少计算量
- 关注生物学变异
- 降低技术噪音影响
Seurat
# 识别高变异基因
pbmc <- FindVariableFeatures(pbmc,
selection.method = "vst",
nfeatures = 2000)
# 查看前10个高变异基因
top10 <- head(VariableFeatures(pbmc), 10)
print(top10)
# 可视化
plot1 <- VariableFeaturePlot(pbmc)
plot2 <- LabelPoints(plot = plot1, points = top10, repel = TRUE)
plot2
Scanpy
# 识别高变异基因
sc.pp.highly_variable_genes(adata,
min_mean=0.0125,
max_mean=3,
min_disp=0.5)
# 可视化
sc.pl.highly_variable_genes(adata)
# 只保留高变异基因用于下游分析
adata = adata[:, adata.var.highly_variable]
数据缩放
Seurat
# 缩放数据
all.genes <- rownames(pbmc)
pbmc <- ScaleData(pbmc, features = all.genes)
# 或只缩放高变异基因
pbmc <- ScaleData(pbmc)
Scanpy
# 回归协变量并缩放
sc.pp.regress_out(adata, ['total_counts', 'pct_counts_mt'])
# 缩放到单位方差
sc.pp.scale(adata, max_value=10)
降维分析
PCA(主成分分析)
Seurat
# 运行 PCA
pbmc <- RunPCA(pbmc,
features = VariableFeatures(object = pbmc),
npcs = 50)
# 查看 PCA 结果
print(pbmc[["pca"]], dims = 1:5, nfeatures = 5)
# 可视化
VizDimLoadings(pbmc, dims = 1:2, reduction = "pca")
DimPlot(pbmc, reduction = "pca")
DimHeatmap(pbmc, dims = 1:15, cells = 500, balanced = TRUE)
Scanpy
# 运行 PCA
sc.tl.pca(adata, svd_solver='arpack')
# 可视化
sc.pl.pca(adata, color='CST3')
sc.pl.pca_variance_ratio(adata, log=True)
确定 PC 数量
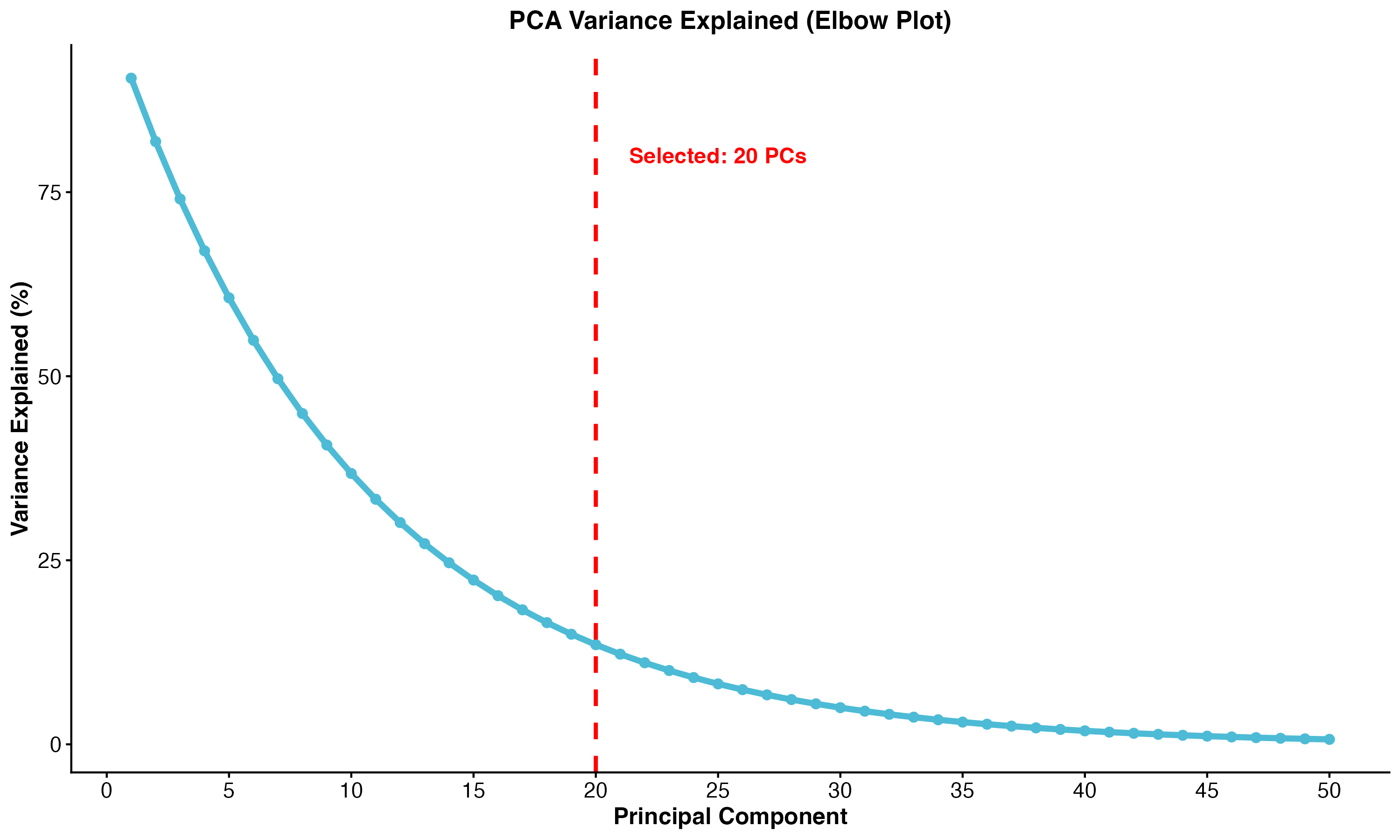
图 5:PCA 方差解释图(Elbow Plot)。展示了每个主成分解释的方差比例,红色虚线标注了选择的 PC 数量(20 个)。
Elbow Plot
# Seurat
ElbowPlot(pbmc, ndims = 50)
# Scanpy
sc.pl.pca_variance_ratio(adata, n_pcs=50)
建议: 选择 elbow 点之前的 PC 数量,通常 15-30 个 PC。
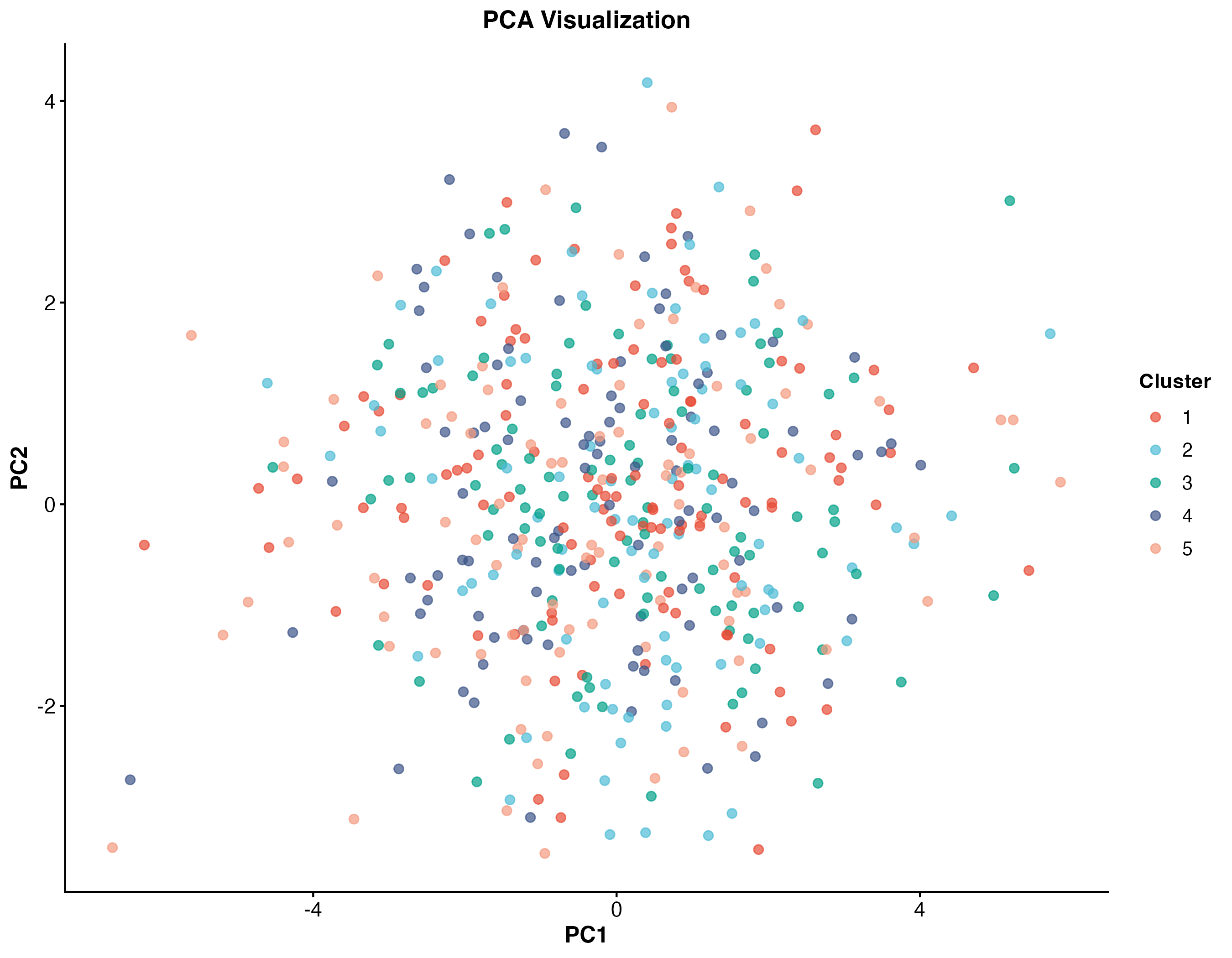
图 6:PCA 散点图。展示了前两个主成分(PC1 和 PC2)的细胞分布,不同颜色代表不同的聚类。
聚类分析
构建邻居图
Seurat
# 构建 KNN 图
pbmc <- FindNeighbors(pbmc, dims = 1:20)
# 聚类
pbmc <- FindClusters(pbmc, resolution = 0.5)
# 查看聚类结果
head(Idents(pbmc), 5)
Scanpy
# 构建邻居图
sc.pp.neighbors(adata, n_neighbors=10, n_pcs=40)
# Leiden 聚类(推荐)
sc.tl.leiden(adata, resolution=0.5)
# 或 Louvain 聚类
sc.tl.louvain(adata, resolution=0.5)
Resolution 参数
- 低 resolution (0.1-0.5): 少量大簇
- 中 resolution (0.5-1.0): 适中数量的簇
- 高 resolution (1.0-2.0): 大量小簇
建议: 从 0.5 开始,根据生物学知识调整。
非线性降维可视化
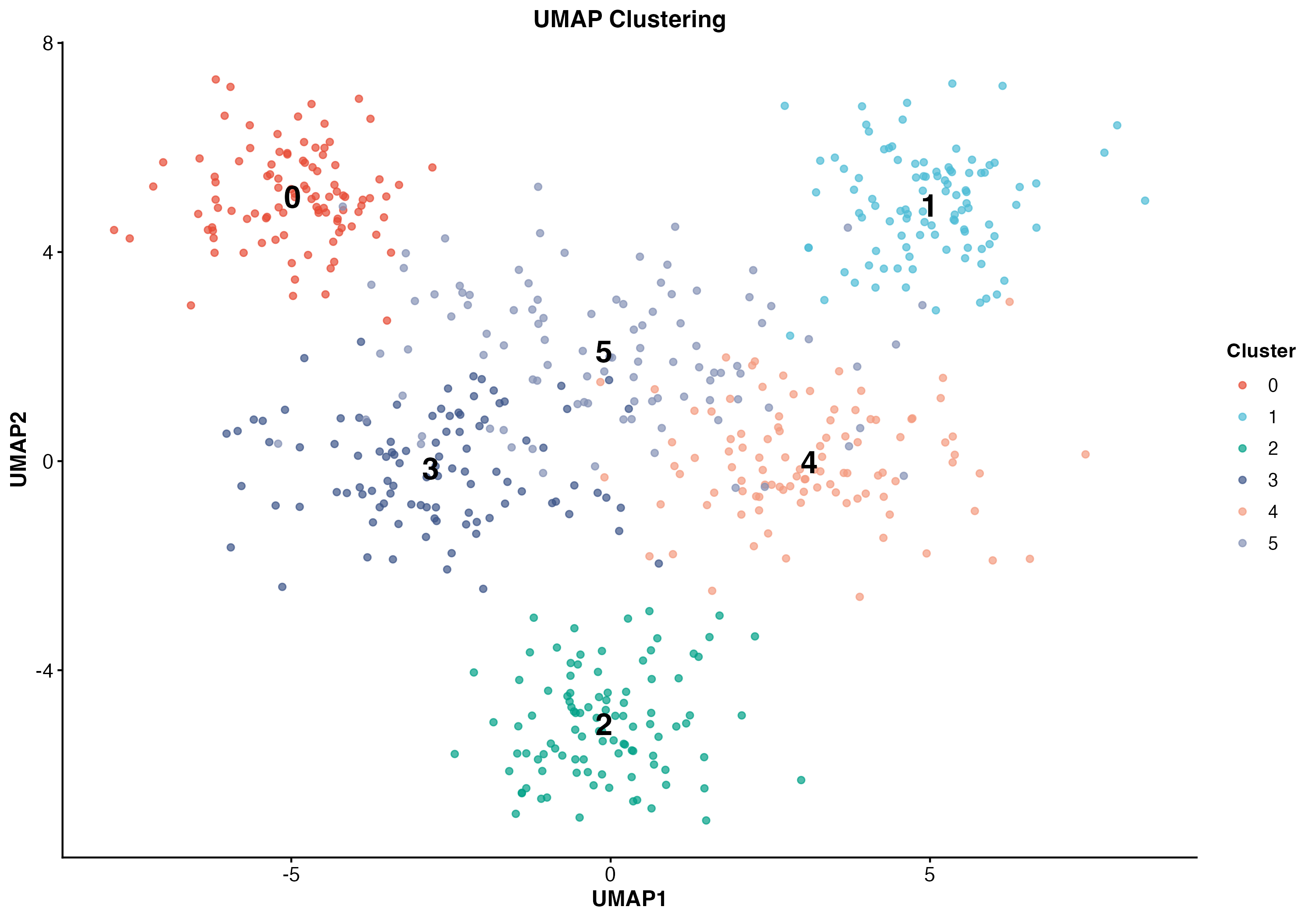
图 7:UMAP 聚类可视化。展示了 6 个细胞簇在 UMAP 空间中的分布,数字标注了簇的编号。
UMAP
Seurat
# 运行 UMAP
pbmc <- RunUMAP(pbmc, dims = 1:20)
# 可视化
DimPlot(pbmc, reduction = "umap", label = TRUE)
# 按基因表达着色
FeaturePlot(pbmc, features = c("MS4A1", "CD79A", "CD3D", "CD8A"))
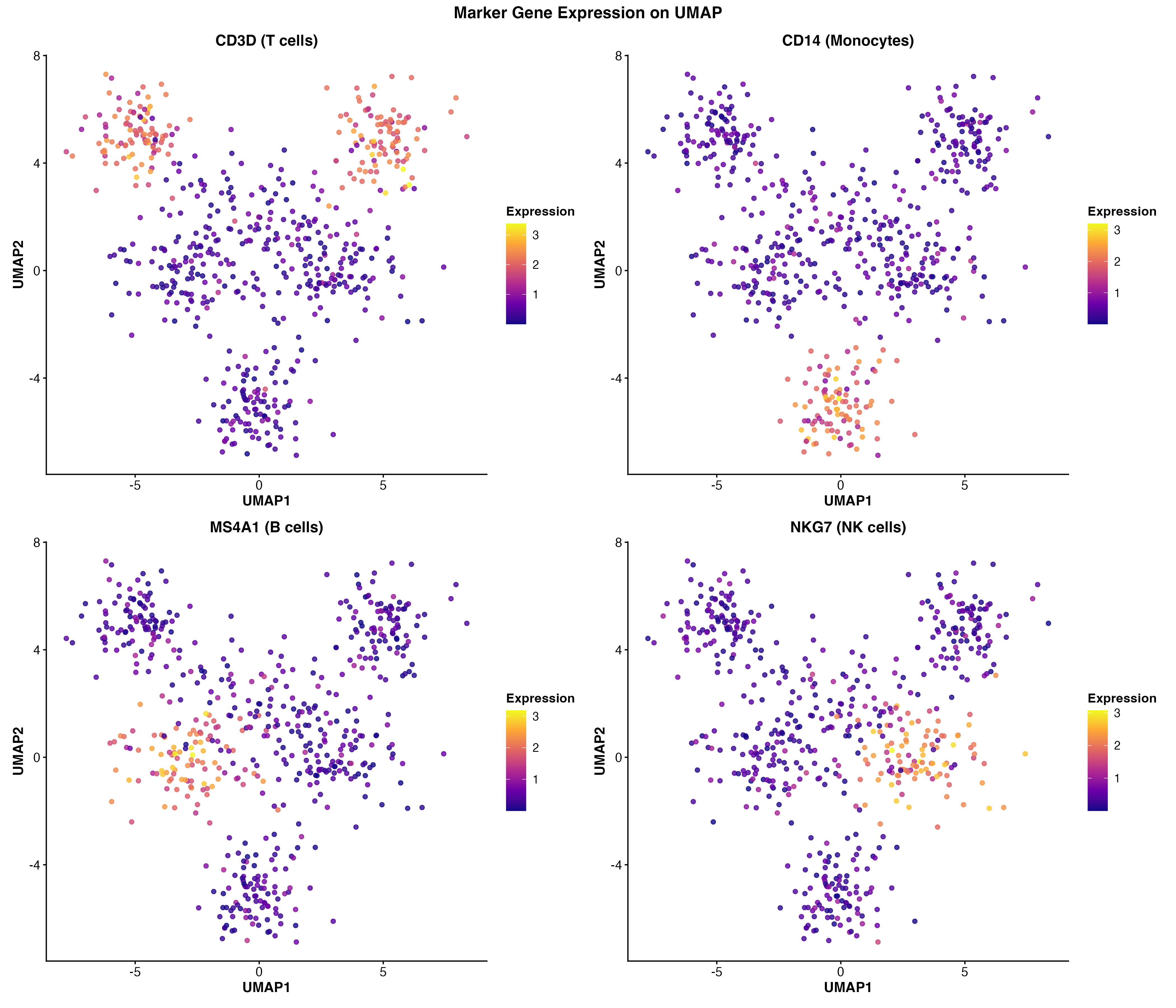
图 8:UMAP 上的标志基因表达。展示了 CD3D(T 细胞)、CD14(单核细胞)、MS4A1(B 细胞)和 NKG7(NK 细胞)的表达模式。
Scanpy
# 运行 UMAP
sc.tl.umap(adata)
# 可视化
sc.pl.umap(adata, color=['leiden', 'CST3', 'NKG7'])
t-SNE(可选)
# Seurat
pbmc <- RunTSNE(pbmc, dims = 1:20)
DimPlot(pbmc, reduction = "tsne")
# Scanpy
sc.tl.tsne(adata, n_pcs=40)
sc.pl.tsne(adata, color='leiden')
寻找标志基因
Seurat
# 寻找所有簇的标志基因
pbmc.markers <- FindAllMarkers(pbmc,
only.pos = TRUE,
min.pct = 0.25,
logfc.threshold = 0.25)
# 查看前5个标志基因
pbmc.markers %>%
group_by(cluster) %>%
slice_max(n = 5, order_by = avg_log2FC)
# 寻找特定簇的标志基因
cluster0.markers <- FindMarkers(pbmc, ident.1 = 0, min.pct = 0.25)
head(cluster0.markers, n = 5)
# 可视化
VlnPlot(pbmc, features = c("MS4A1", "CD79A"))
FeaturePlot(pbmc, features = c("MS4A1", "GNLY", "CD3E", "CD14"))
# 热图
top10 <- pbmc.markers %>%
group_by(cluster) %>%
top_n(n = 10, wt = avg_log2FC)
DoHeatmap(pbmc, features = top10$gene) + NoLegend()
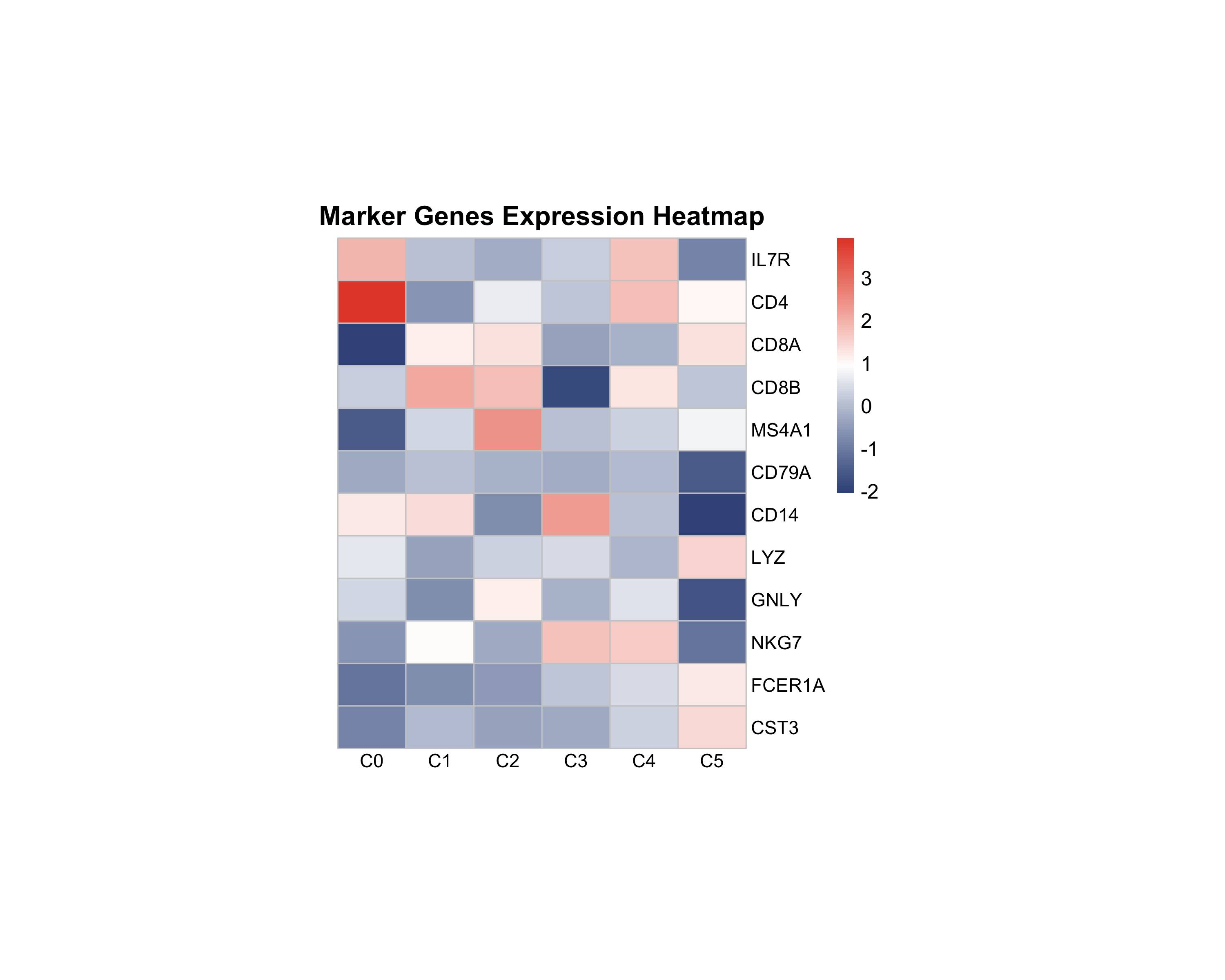
图 9:标志基因表达热图。展示了不同细胞类型的特征标志基因表达模式,每列代表一个细胞簇。
Scanpy
# 寻找标志基因
sc.tl.rank_genes_groups(adata, 'leiden', method='wilcoxon')
# 可视化
sc.pl.rank_genes_groups(adata, n_genes=25, sharey=False)
# 查看结果
result = adata.uns['rank_genes_groups']
groups = result['names'].dtype.names
pd.DataFrame(
{group + '_' + key[:1]: result[key][group]
for group in groups for key in ['names', 'pvals']}).head(5)
# 点图
sc.pl.dotplot(adata,
var_names=['MS4A1', 'CD79A', 'CD3D', 'CD8A'],
groupby='leiden')
# 热图
sc.pl.rank_genes_groups_heatmap(adata, n_genes=10, groupby='leiden')
细胞类型注释
手动注释
基于已知的标志基因:
PBMC 细胞类型标志基因
| 细胞类型 | 标志基因 |
|---|---|
| CD4+ T 细胞 | IL7R, CD4 |
| CD8+ T 细胞 | CD8A, CD8B |
| B 细胞 | MS4A1 (CD20), CD79A |
| NK 细胞 | GNLY, NKG7 |
| 单核细胞 | CD14, LYZ |
| 树突状细胞 | FCER1A, CST3 |
| 巨核细胞 | PPBP |
Seurat 注释
# 定义细胞类型
new.cluster.ids <- c(
"Naive CD4 T", "CD14+ Mono", "Memory CD4 T",
"B", "CD8 T", "FCGR3A+ Mono",
"NK", "DC", "Platelet"
)
names(new.cluster.ids) <- levels(pbmc)
pbmc <- RenameIdents(pbmc, new.cluster.ids)
# 可视化
DimPlot(pbmc, reduction = "umap", label = TRUE, pt.size = 0.5) + NoLegend()
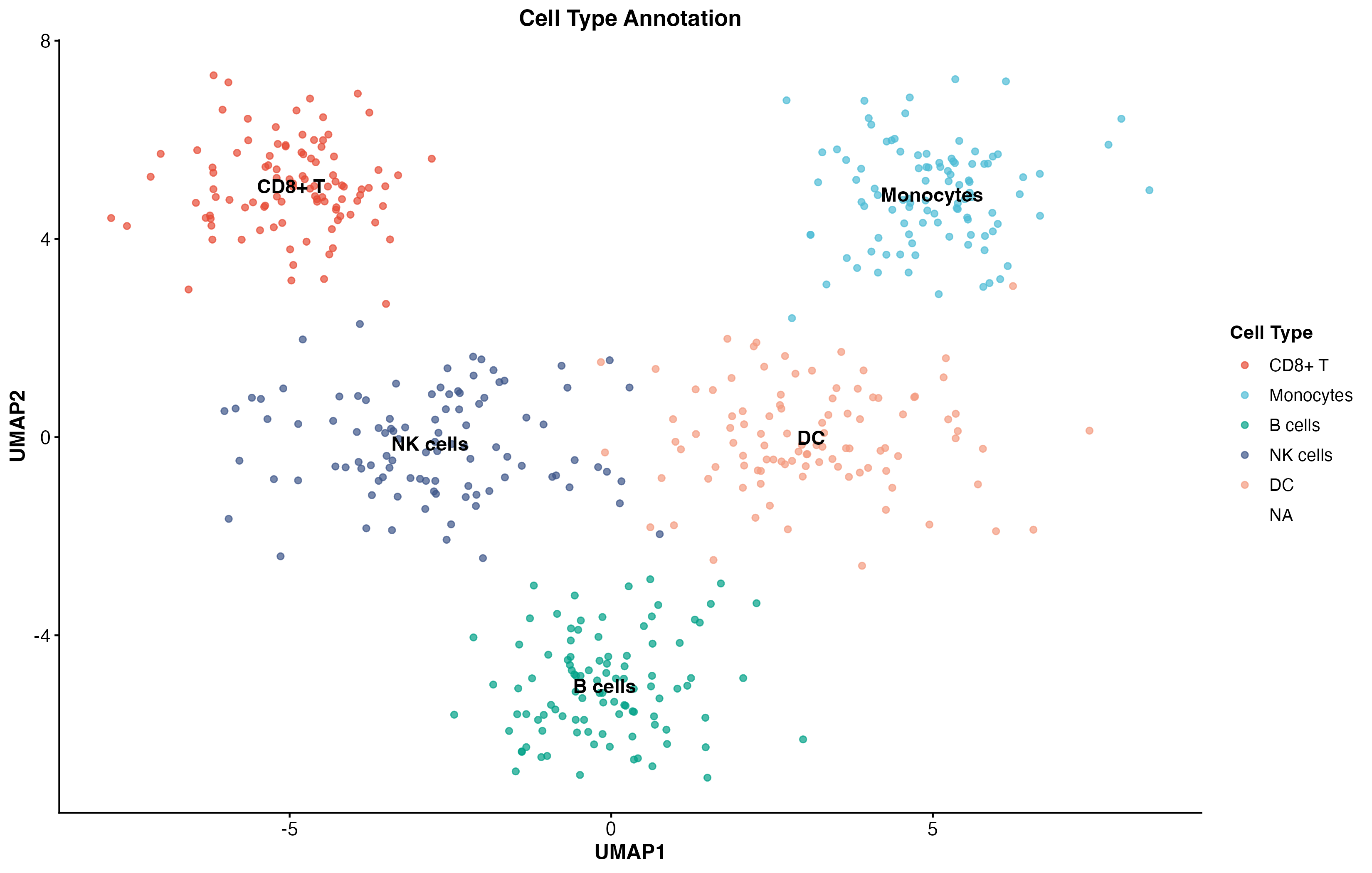
图 10:细胞类型注释 UMAP 图。展示了 6 种主要细胞类型在 UMAP 空间中的分布。
Scanpy 注释
# 定义细胞类型
cluster2annotation = {
'0': 'CD4 T',
'1': 'CD14 Monocytes',
'2': 'B',
'3': 'CD8 T',
'4': 'NK',
'5': 'FCGR3A Monocytes',
'6': 'Dendritic',
'7': 'Megakaryocytes'
}
adata.obs['cell_type'] = adata.obs['leiden'].map(cluster2annotation)
# 可视化
sc.pl.umap(adata, color='cell_type', legend_loc='on data')
自动注释(SingleR)
library(SingleR)
library(celldex)
# 加载参考数据集
ref <- celldex::HumanPrimaryCellAtlasData()
# 转换为 SingleCellExperiment
sce <- as.SingleCellExperiment(pbmc)
# 运行 SingleR
pred <- SingleR(test = sce,
ref = ref,
labels = ref$label.main)
# 添加到 Seurat 对象
pbmc$singler_labels <- pred$labels
# 可视化
DimPlot(pbmc, reduction = "umap", group.by = "singler_labels")
差异表达分析
比较两组细胞
# Seurat
# 比较 CD4 T 和 CD8 T 细胞
cd4_vs_cd8 <- FindMarkers(pbmc,
ident.1 = "CD4 T",
ident.2 = "CD8 T")
head(cd4_vs_cd8)
# Scanpy
# 比较两个簇
sc.tl.rank_genes_groups(adata,
'cell_type',
groups=['CD4 T'],
reference='CD8 T',
method='wilcoxon')
sc.pl.rank_genes_groups(adata)
功能富集分析
GO 富集分析
library(clusterProfiler)
library(org.Hs.eg.db)
# 获取簇0的标志基因
cluster0_genes <- pbmc.markers %>%
filter(cluster == 0 & p_val_adj < 0.05) %>%
pull(gene)
# 转换基因 ID
gene_ids <- bitr(cluster0_genes,
fromType = "SYMBOL",
toType = "ENTREZID",
OrgDb = org.Hs.eg.db)
# GO 富集
ego <- enrichGO(gene = gene_ids$ENTREZID,
OrgDb = org.Hs.eg.db,
ont = "BP",
pAdjustMethod = "BH",
pvalueCutoff = 0.05,
qvalueCutoff = 0.05)
# 可视化
barplot(ego, showCategory=10)
dotplot(ego, showCategory=10)
KEGG 通路富集
# KEGG 富集
kk <- enrichKEGG(gene = gene_ids$ENTREZID,
organism = 'hsa',
pvalueCutoff = 0.05)
# 可视化
dotplot(kk, showCategory=10)
保存结果
Seurat
# 保存 Seurat 对象
saveRDS(pbmc, file = "pbmc_analyzed.rds")
# 导出表达矩阵
write.csv(as.matrix(pbmc@assays$RNA@counts),
file = "expression_matrix.csv")
# 导出元数据
write.csv(pbmc@meta.data, file = "metadata.csv")
# 导出标志基因
write.csv(pbmc.markers, file = "marker_genes.csv")
Scanpy
# 保存 AnnData 对象
adata.write('pbmc_analyzed.h5ad')
# 导出为 CSV
adata.obs.to_csv('metadata.csv')
adata.var.to_csv('genes.csv')
最佳实践
1. QC 阈值设置
- 根据数据分布设置阈值
- 不同组织/细胞类型阈值不同
- 保守过滤,避免丢失真实细胞
2. 标准化方法选择
- LogNormalize: 简单快速
- SCTransform: 更好地处理技术噪音(推荐)
3. 聚类参数
- 尝试多个 resolution 值
- 结合生物学知识判断
- 使用 clustree 包可视化不同 resolution
4. 细胞类型注释
- 结合多个标志基因
- 使用自动注释工具辅助
- 查阅文献验证
常见问题
问题 1: 聚类结果不理想
解决方案:
- 调整 resolution 参数
- 增加或减少 PC 数量
- 检查 QC 是否充分
- 尝试不同的聚类算法
问题 2: 找不到明显的标志基因
解决方案:
- 降低 logfc.threshold
- 增加细胞数量
- 检查数据质量
- 考虑细胞亚型过于相似
问题 3: 双细胞问题
解决方案:
- 使用 DoubletFinder (Seurat)
- 使用 Scrublet (Scanpy)
- 严格的 QC 过滤
下一步
完成质量控制和聚类后,可以进行:
- 数据整合 - 合并多个样本
- 轨迹推断 - 研究细胞分化
- 细胞通讯 - 分析细胞间相互作用
- 差异分析 - 比较不同条件
继续学习:模块04:多样本数据整合