模块02:原始数据处理与 Cell Ranger
本模块将介绍如何使用 Cell Ranger 处理 10x Genomics 单细胞 RNA 测序的原始数据。
学习目标
完成本模块后,你将能够:
- 理解单细胞测序原始数据的格式和结构
- 掌握 Cell Ranger 的基本使用方法
- 进行序列比对和基因定量
- 理解输出文件的含义和用途
- 评估数据质量
前置知识
- Linux 命令行基础
- 基因组学基本概念
- 测序技术基础知识
单细胞测序数据概述
10x Genomics 技术
10x Genomics 是目前最流行的单细胞转录组测序平台之一。
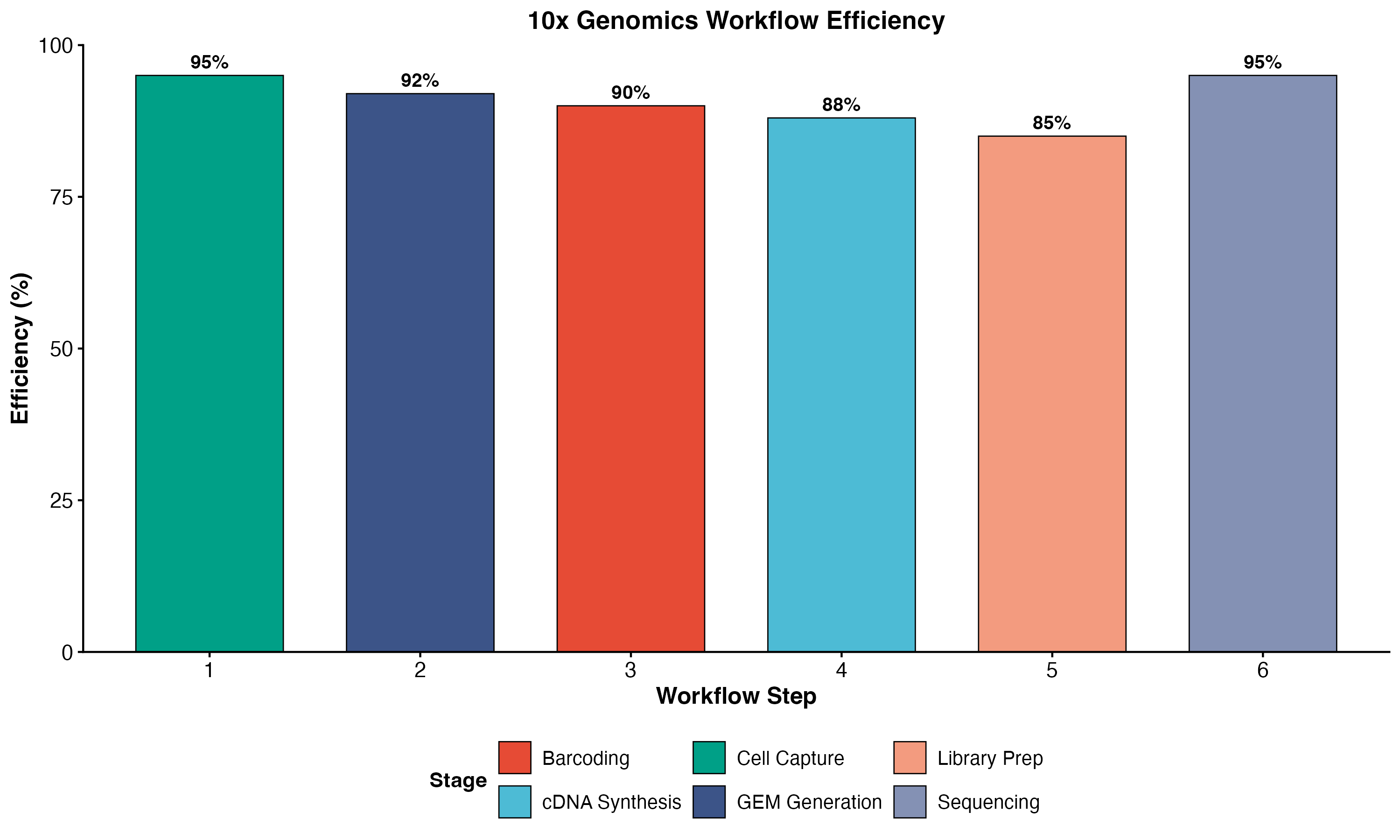
图 1:10x Genomics 工作流程各阶段效率。展示了从细胞捕获到测序的完整流程及各步骤的效率。
技术特点:
- 基于微流控技术
- 使用 Gel Beads in Emulsion (GEM)
- 每个细胞有唯一的 barcode
- 每个 mRNA 分子有唯一的 UMI (Unique Molecular Identifier)
数据结构
FASTQ 文件:
@序列ID
ATCGATCGATCG...
+
IIIIIIIIIIII...
文件组成:
R1.fastq.gz: Read 1 - 包含 barcode 和 UMIR2.fastq.gz: Read 2 - 包含 cDNA 序列I1.fastq.gz: Index - 样本索引(可选)
Barcode 和 UMI
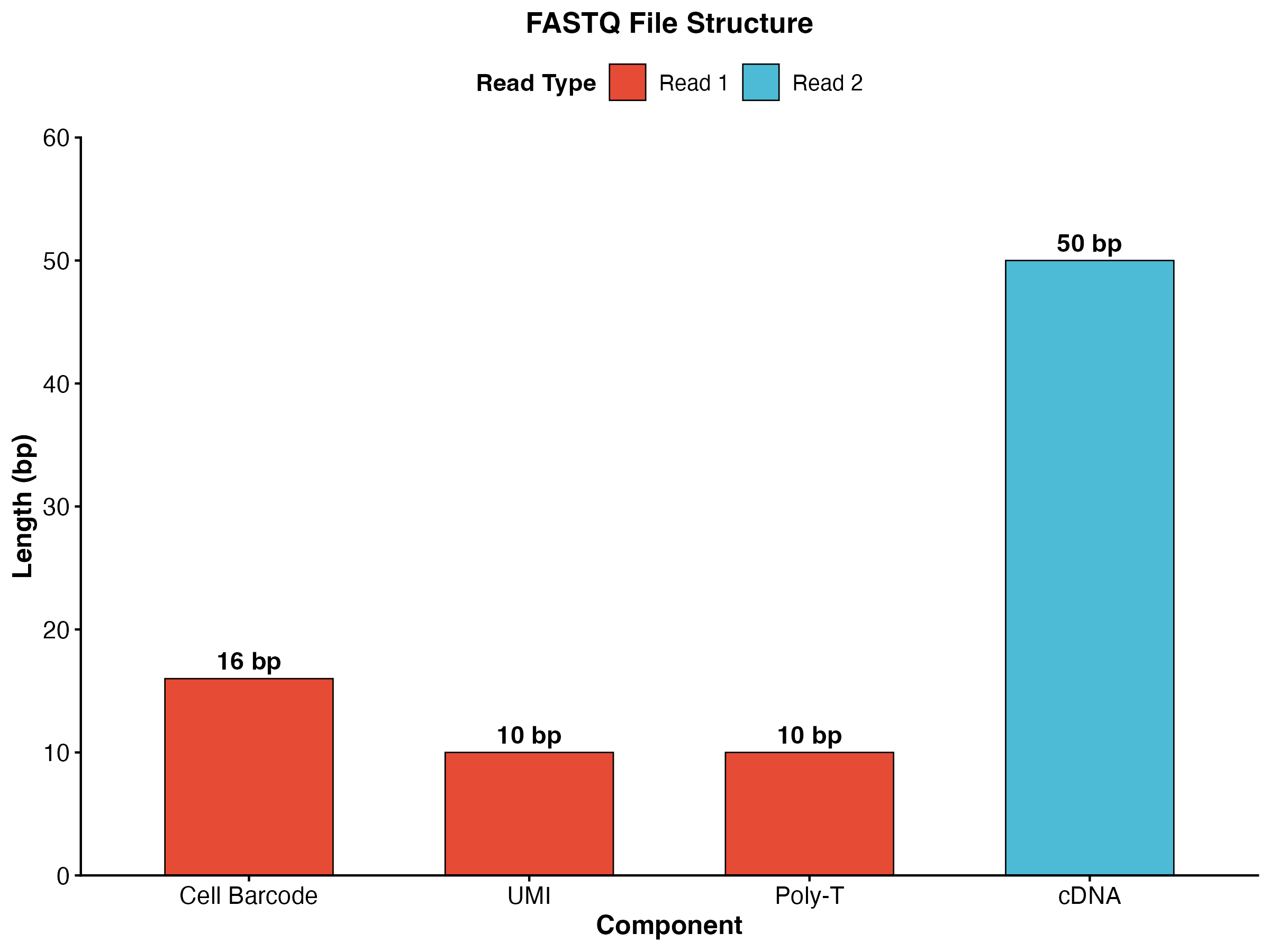
图 2:FASTQ 文件结构示意图。展示了 Read 1 和 Read 2 中各组成部分的长度和位置。
Read 1 结构:
[16bp Cell Barcode][10bp UMI][Poly-T]
Read 2 结构:
[cDNA sequence]
作用:
- Cell Barcode: 标识细胞来源
- UMI: 标识原始 mRNA 分子,用于去重
- cDNA: 基因序列信息
Cell Ranger 简介
什么是 Cell Ranger?
Cell Ranger 是 10x Genomics 开发的官方分析软件,用于处理单细胞测序数据。
主要功能:
- FASTQ 文件处理
- 序列比对到参考基因组
- Barcode 识别和校正
- UMI 计数
- 生成基因表达矩阵
- 质量控制报告
系统要求
硬件要求:
- CPU: 8+ 核心
- 内存: 64GB+ RAM
- 存储: 1TB+ 可用空间
软件要求:
- Linux 操作系统
- Cell Ranger 软件
- 参考基因组
安装 Cell Ranger
下载
# 访问 10x Genomics 官网下载
# https://support.10xgenomics.com/single-cell-gene-expression/software/downloads/latest
# 或使用 wget
wget -O cellranger-8.0.0.tar.gz \
"https://cf.10xgenomics.com/releases/cell-exp/cellranger-8.0.0.tar.gz"
安装
# 解压
tar -xzvf cellranger-8.0.0.tar.gz
# 添加到 PATH
export PATH=/path/to/cellranger-8.0.0:$PATH
# 验证安装
cellranger --version
下载参考基因组
# 人类参考基因组 (GRCh38)
wget https://cf.10xgenomics.com/supp/cell-exp/refdata-gex-GRCh38-2024-A.tar.gz
# 小鼠参考基因组 (mm10)
wget https://cf.10xgenomics.com/supp/cell-exp/refdata-gex-mm10-2024-A.tar.gz
# 解压
tar -xzvf refdata-gex-GRCh38-2024-A.tar.gz
Cell Ranger 工作流程
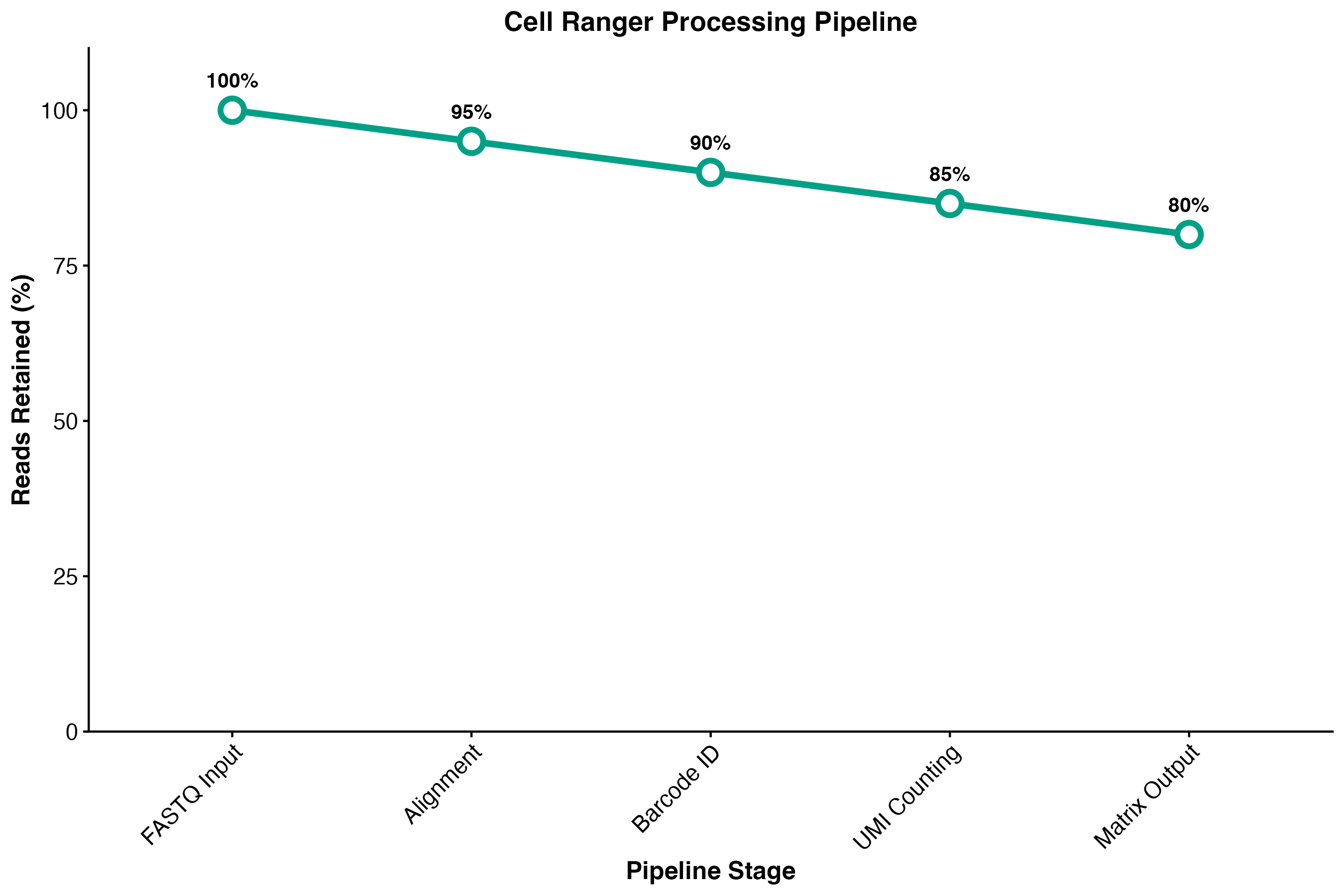
图 3:Cell Ranger 处理流程各阶段。展示了从 FASTQ 输入到表达矩阵输出的完整流程及每个阶段保留的 reads 比例。
完整流程
FASTQ 文件
↓
cellranger count
↓
├─ 序列比对 (STAR)
├─ Barcode 识别
├─ UMI 计数
└─ 基因定量
↓
输出文件
├─ 表达矩阵
├─ BAM 文件
└─ 质量报告
主要命令
- cellranger count: 主要分析流程
- cellranger aggr: 合并多个样本
- cellranger reanalyze: 重新分析
- cellranger mkref: 构建自定义参考基因组
使用 Cell Ranger Count
基本用法
cellranger count \
--id=sample_01 \
--transcriptome=/path/to/refdata-gex-GRCh38-2024-A \
--fastqs=/path/to/fastq_folder \
--sample=sample_name \
--localcores=8 \
--localmem=64
参数说明
| 参数 | 说明 | 必需 |
|---|---|---|
--id | 输出目录名称 | ✅ |
--transcriptome | 参考基因组路径 | ✅ |
--fastqs | FASTQ 文件目录 | ✅ |
--sample | 样本名称 | ✅ |
--localcores | CPU 核心数 | ❌ |
--localmem | 内存大小 (GB) | ❌ |
--expect-cells | 预期细胞数 | ❌ |
--chemistry | 化学试剂版本 | ❌ |
实际示例
# 示例 1: 基本分析
cellranger count \
--id=PBMC_sample1 \
--transcriptome=/data/refdata-gex-GRCh38-2024-A \
--fastqs=/data/fastq/PBMC \
--sample=PBMC_1 \
--localcores=16 \
--localmem=128
# 示例 2: 指定预期细胞数
cellranger count \
--id=tumor_sample \
--transcriptome=/data/refdata-gex-GRCh38-2024-A \
--fastqs=/data/fastq/tumor \
--sample=tumor_01 \
--expect-cells=5000 \
--localcores=16 \
--localmem=128
# 示例 3: 多个 FASTQ 目录
cellranger count \
--id=multi_lane \
--transcriptome=/data/refdata-gex-GRCh38-2024-A \
--fastqs=/data/lane1,/data/lane2 \
--sample=sample_A \
--localcores=16 \
--localmem=128
输出文件结构
目录结构
sample_01/
├── outs/
│ ├── web_summary.html # 质量报告(重要)
│ ├── metrics_summary.csv # 统计指标
│ ├── filtered_feature_bc_matrix/ # 过滤后的表达矩阵(重要)
│ │ ├── barcodes.tsv.gz
│ │ ├── features.tsv.gz
│ │ └── matrix.mtx.gz
│ ├── raw_feature_bc_matrix/ # 原始表达矩阵
│ │ ├── barcodes.tsv.gz
│ │ ├── features.tsv.gz
│ │ └── matrix.mtx.gz
│ ├── possorted_genome_bam.bam # 比对文件
│ ├── possorted_genome_bam.bam.bai
│ ├── filtered_feature_bc_matrix.h5 # HDF5 格式(重要)
│ ├── raw_feature_bc_matrix.h5
│ ├── molecule_info.h5
│ └── cloupe.cloupe # Loupe Browser 文件
└── SC_RNA_COUNTER_CS/
└── ... # 中间文件
重要文件说明
1. web_summary.html
内容:
- 测序质量统计
- 细胞数量
- 基因检测数
- UMI 计数
- 比对率
- 可视化图表
查看方式:
# 在浏览器中打开
firefox sample_01/outs/web_summary.html
2. filtered_feature_bc_matrix/
包含三个文件:
barcodes.tsv.gz: 细胞 barcode 列表
AAACCCAAGAAACACT-1
AAACCCAAGAAACCAT-1
AAACCCAAGAAACCGC-1
...
features.tsv.gz: 基因信息
ENSG00000243485 MIR1302-2HG Gene Expression
ENSG00000237613 FAM138A Gene Expression
ENSG00000186092 OR4F5 Gene Expression
...
matrix.mtx.gz: 表达矩阵(稀疏矩阵格式)
%%MatrixMarket matrix coordinate integer general
33538 4000 10000000
1 1 5
1 2 3
2 1 8
...
3. filtered_feature_bc_matrix.h5
HDF5 格式的表达矩阵,包含所有信息,便于读取。
质量控制指标
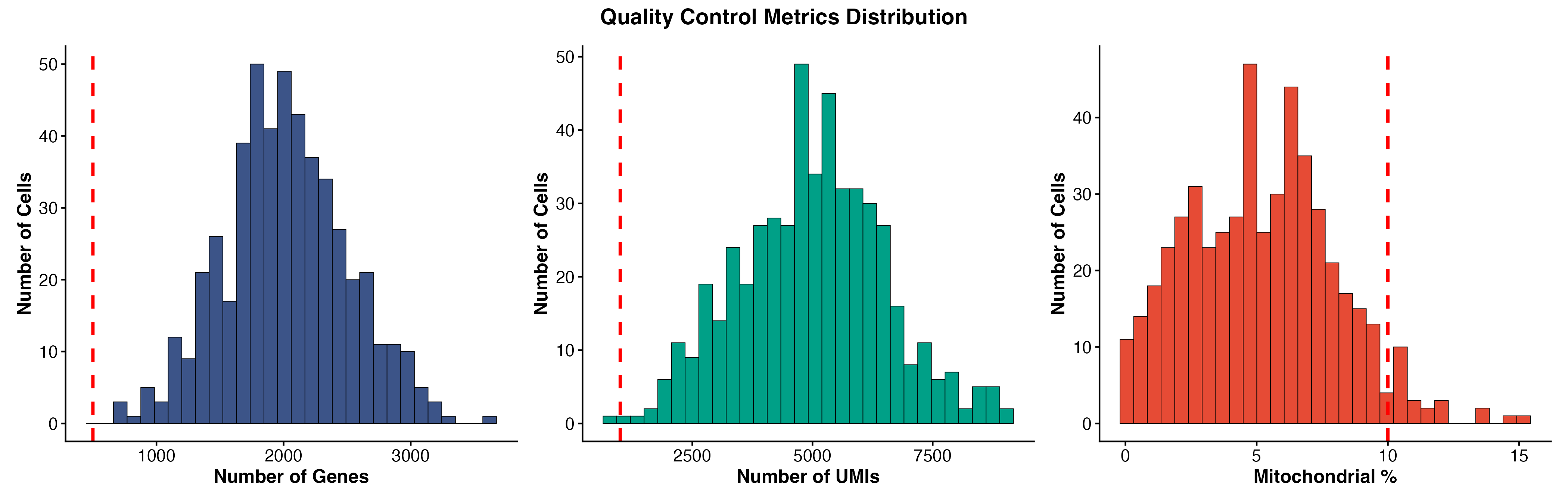
图 4:质量控制指标分布。展示了基因数、UMI 数和线粒体基因比例的分布情况,红色虚线表示质量控制阈值。
关键指标
1. 细胞数量 (Estimated Number of Cells)
含义: 检测到的细胞数量
正常范围:
- 取决于上样量
- 通常 1,000 - 10,000 个细胞
异常情况:
- 过低: 可能是细胞死亡、上样量不足
- 过高: 可能包含双细胞
2. 平均每个细胞的基因数 (Mean Reads per Cell)
含义: 测序深度
正常范围:
- 20,000 - 100,000 reads/cell
建议:
- 最低 10,000 reads/cell
- 更高的深度可以检测更多基因
3. 中位基因数 (Median Genes per Cell)
含义: 每个细胞检测到的基因数量
正常范围:
- 500 - 5,000 基因/细胞
- 取决于细胞类型
异常情况:
- 过低: 细胞质量差、测序深度不足
- 过高: 可能是双细胞
4. 总基因检测数 (Total Genes Detected)
含义: 在所有细胞中检测到的基因总数
正常范围:
- 人类: 15,000 - 25,000 基因
- 小鼠: 15,000 - 25,000 基因
5. 测序饱和度 (Sequencing Saturation)
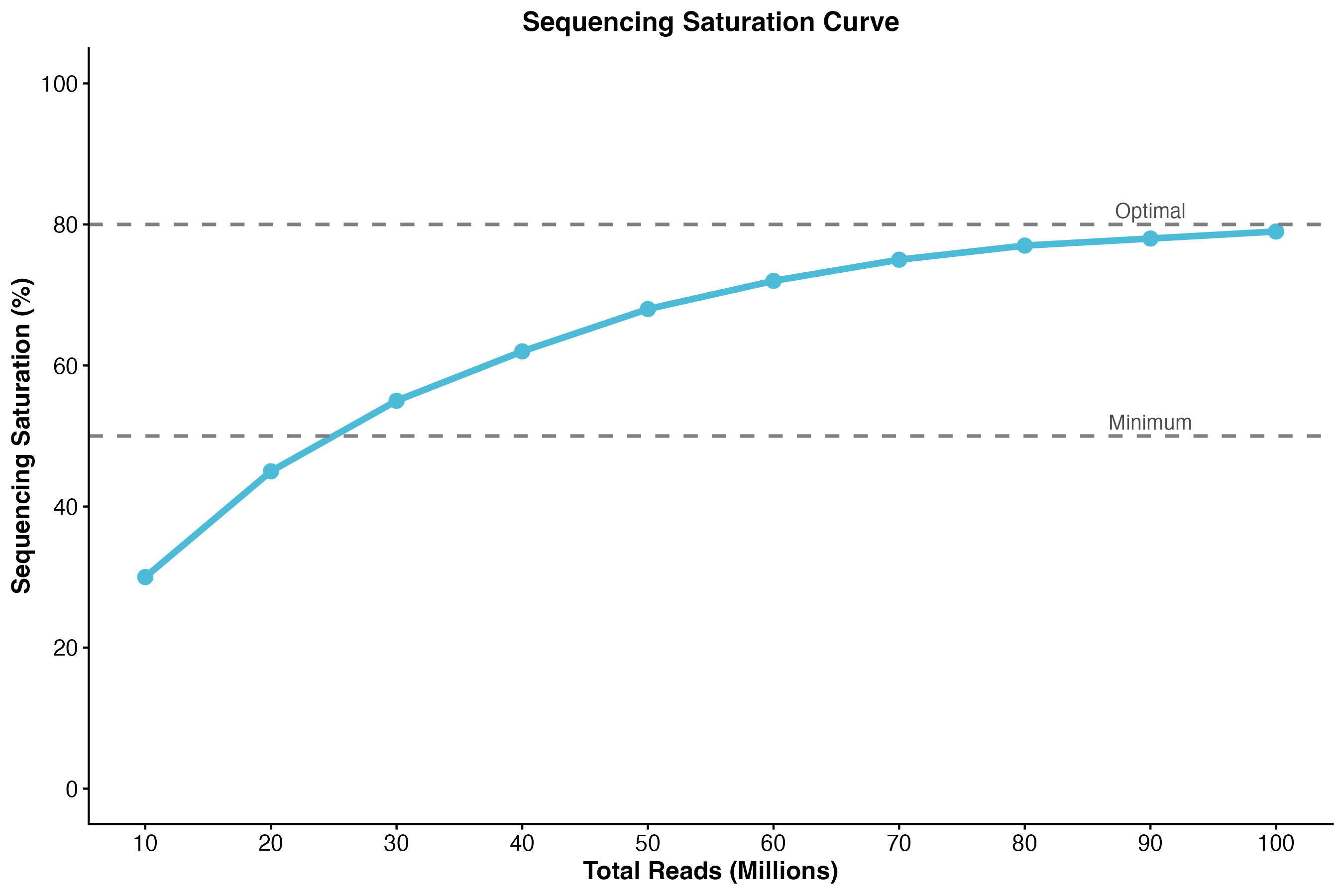
图 5:测序饱和度曲线。展示了测序深度与饱和度的关系,虚线标注了最低要求(50%)和最佳范围(80%)。
含义: 测序深度是否足够
计算公式:
饱和度 = 1 - (unique UMIs / total reads)
正常范围:
- 50% - 80%
解释:
- 低饱和度: 可以增加测序深度
- 高饱和度: 继续测序收益递减
6. 比对率 (Reads Mapped to Genome)
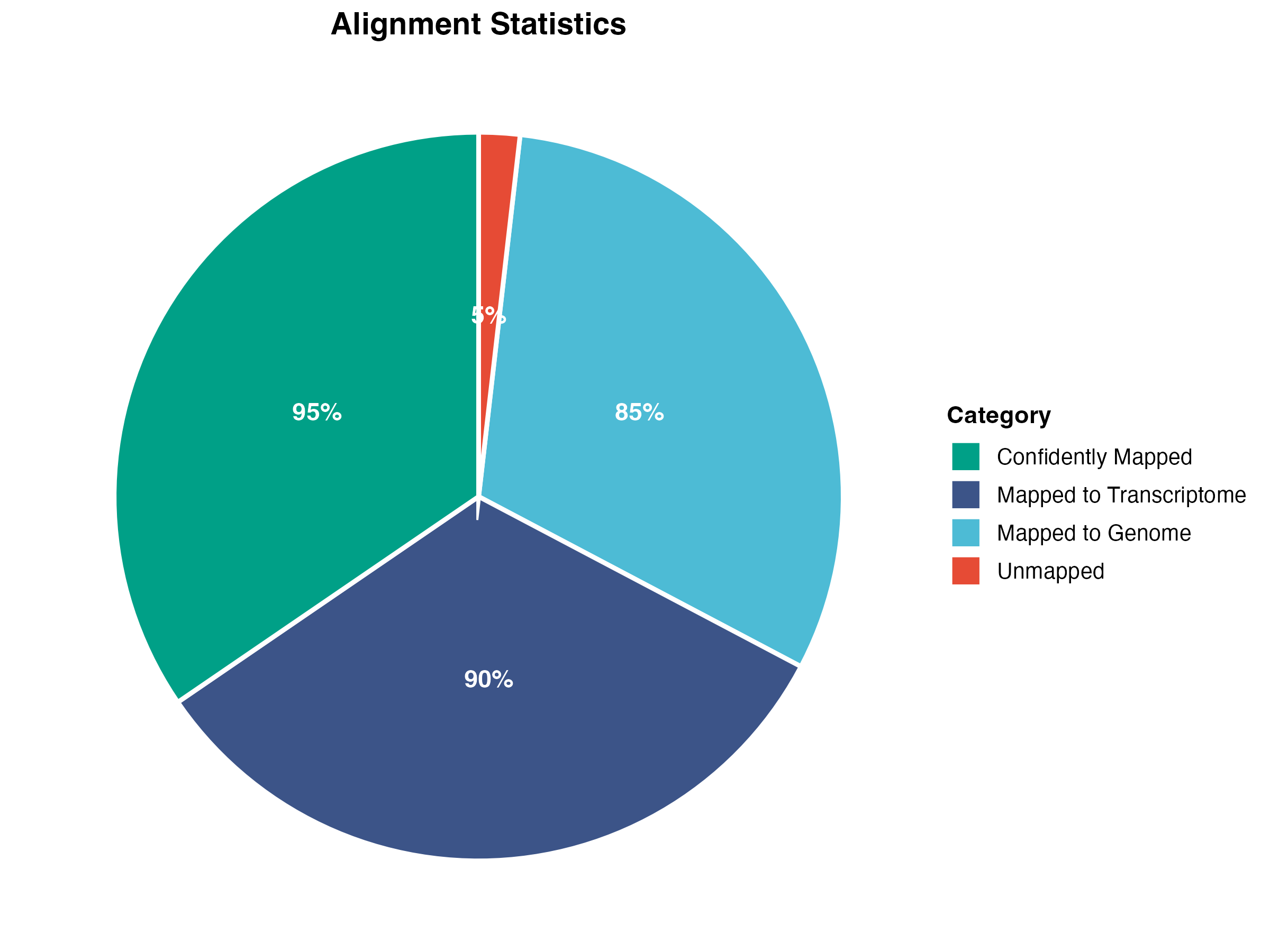
图 6:比对统计饼图。展示了不同类型比对结果的比例分布。
含义: 成功比对到基因组的 reads 比例
正常范围:
-
80%
异常情况:
- 低比对率: 可能是样本污染、参考基因组错误
7. 线粒体基因比例
含义: 线粒体基因表达占比
正常范围:
- < 10%
异常情况:
- 高比例: 细胞质量差、细胞破损
读取数据到 R
Vibe coding 带来极大的便利,能否用好工具需要思想的指引。如果想复现这些分析,建议下载完整脚本学习。
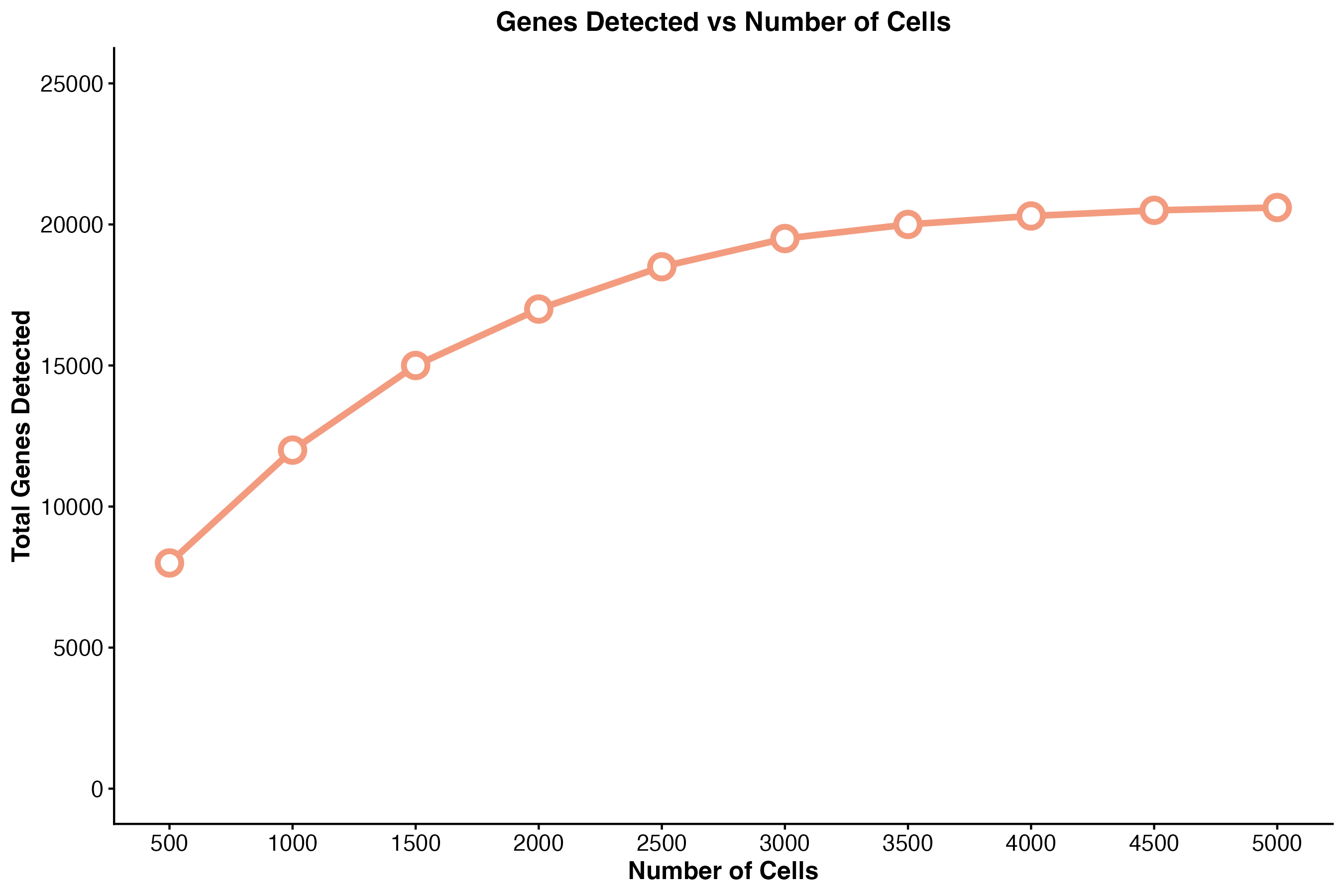
图 7:细胞数量与基因检测关系。展示了随着细胞数量增加,检测到的基因总数的变化趋势。
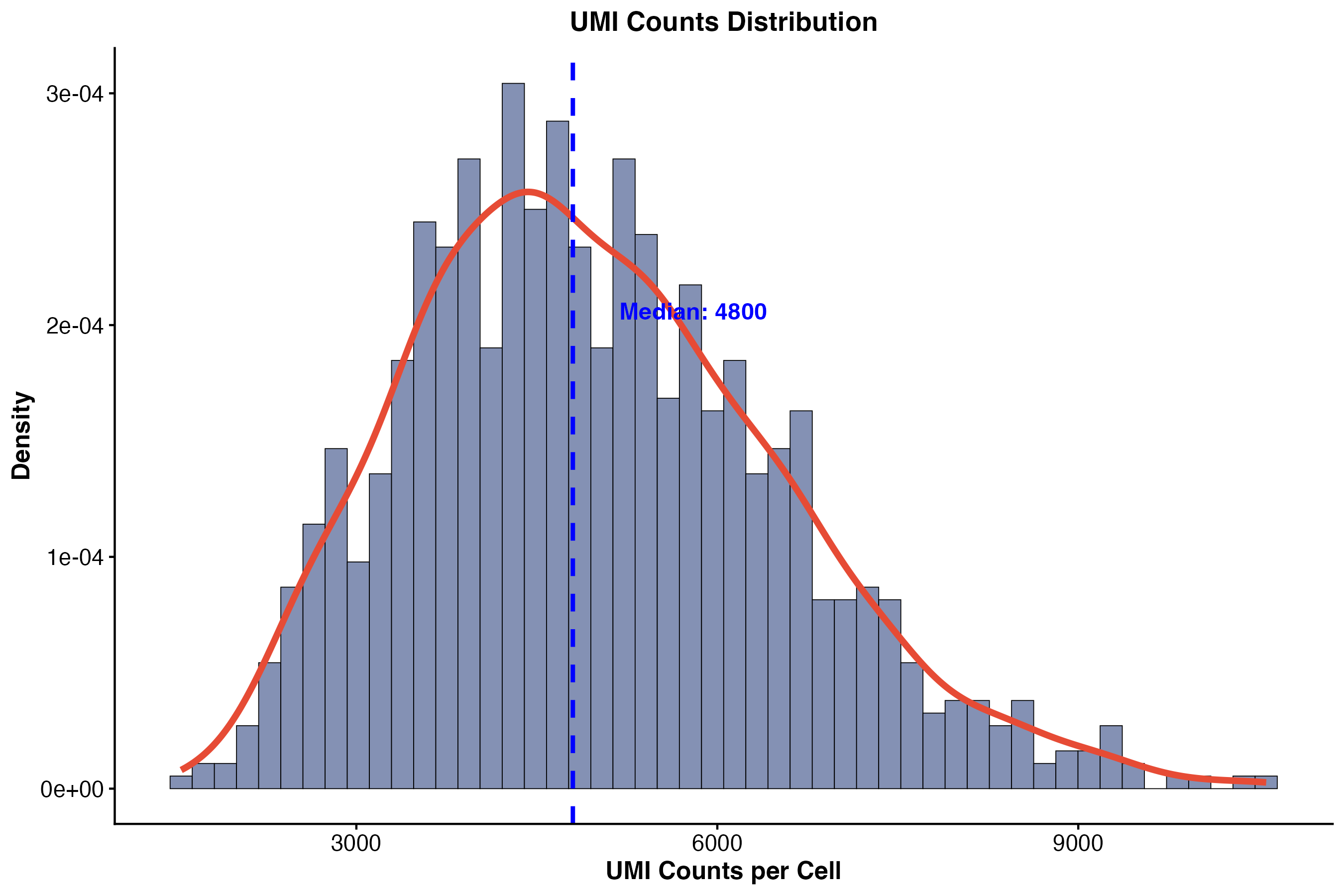
图 8:UMI 计数分布。展示了每个细胞的 UMI 计数分布,蓝色虚线表示中位数。
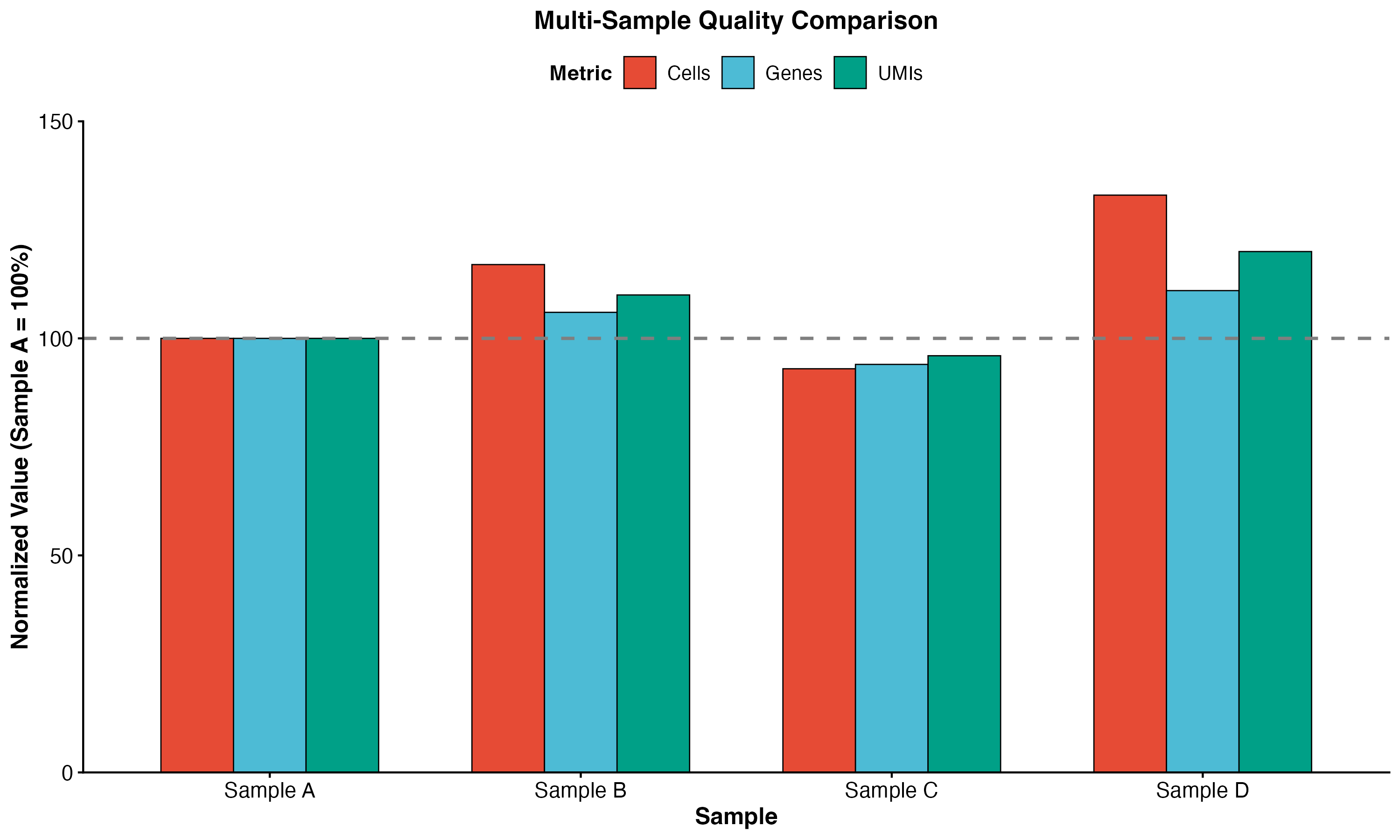
图 9:多样本质量对比。展示了不同样本在细胞数、基因数和 UMI 数三个指标上的标准化比较。
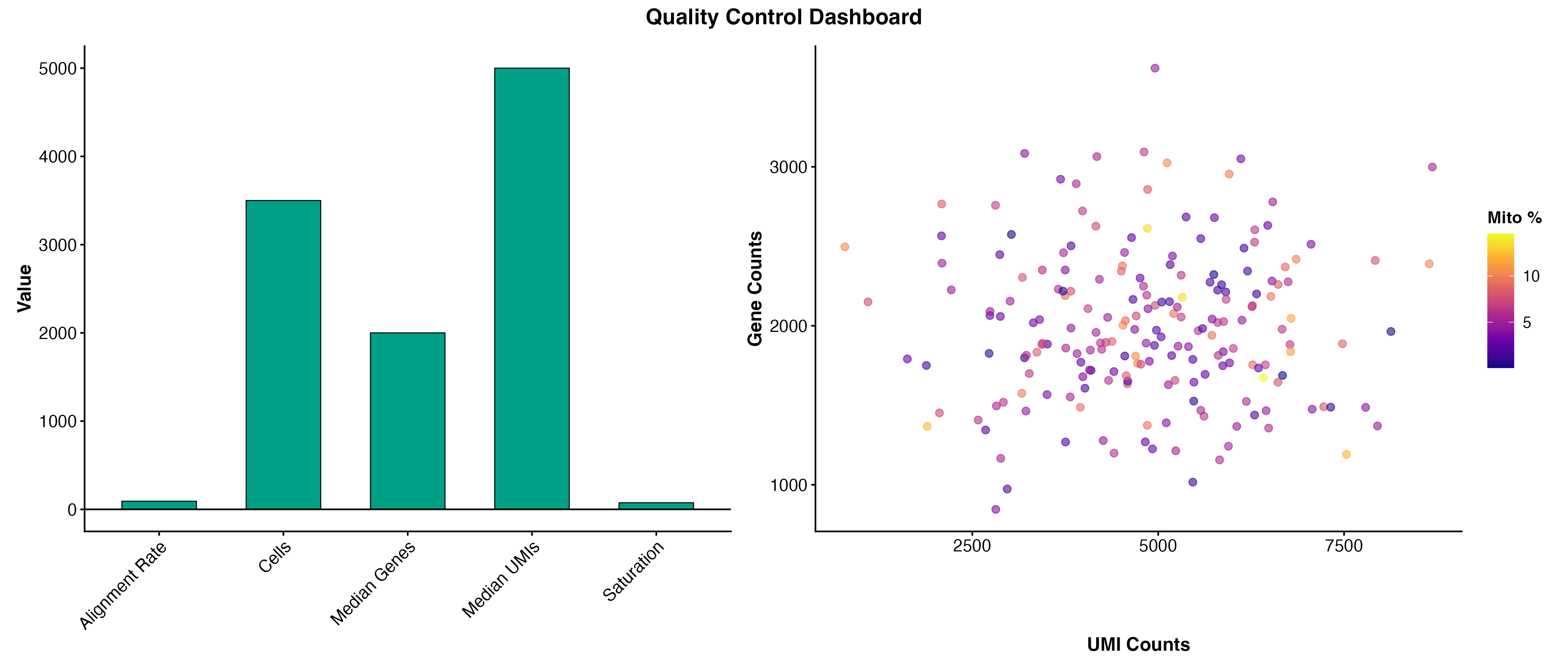
图 10:质量控制仪表盘。综合展示了关键质量指标和细胞质量分布的散点图。
使用 Seurat
library(Seurat)
# 读取 10x 数据
data_dir <- "sample_01/outs/filtered_feature_bc_matrix/"
data <- Read10X(data.dir = data_dir)
# 创建 Seurat 对象
seurat_obj <- CreateSeuratObject(
counts = data,
project = "PBMC",
min.cells = 3,
min.features = 200
)
# 查看对象
seurat_obj
使用 Scanpy (Python)
import scanpy as sc
# 读取 10x 数据
adata = sc.read_10x_mtx(
'sample_01/outs/filtered_feature_bc_matrix/',
var_names='gene_symbols',
cache=True
)
# 查看对象
print(adata)
读取 H5 文件
# R - Seurat
library(Seurat)
data <- Read10X_h5("sample_01/outs/filtered_feature_bc_matrix.h5")
seurat_obj <- CreateSeuratObject(counts = data)
# Python - Scanpy
import scanpy as sc
adata = sc.read_10x_h5('sample_01/outs/filtered_feature_bc_matrix.h5')
常见问题
问题 1: 内存不足
错误信息:
[error] Pipestance failed. Error log at: ...
Out of memory
解决方案:
- 增加
--localmem参数 - 使用更大内存的服务器
- 减少
--localcores参数
问题 2: 细胞数量过低
可能原因:
- 细胞死亡
- 上样量不足
- 实验操作问题
解决方案:
- 检查实验流程
- 调整
--expect-cells参数 - 检查 FASTQ 文件质量
问题 3: 比对率低
可能原因:
- 参考基因组错误
- 样本污染
- 测序质量差
解决方案:
- 确认参考基因组版本
- 检查样本来源
- 查看 FASTQ 质量报告
问题 4: 运行时间过长
优化方法:
- 增加 CPU 核心数
- 使用 SSD 存储
- 检查系统负载
最佳实践
1. 数据组织
project/
├── fastq/
│ ├── sample1/
│ └── sample2/
├── reference/
│ └── refdata-gex-GRCh38-2024-A/
├── analysis/
│ ├── sample1/
│ └── sample2/
└── scripts/
└── run_cellranger.sh
2. 批处理脚本
#!/bin/bash
# run_cellranger.sh
SAMPLES=("sample1" "sample2" "sample3")
TRANSCRIPTOME="/data/refdata-gex-GRCh38-2024-A"
FASTQ_DIR="/data/fastq"
for SAMPLE in "${SAMPLES[@]}"; do
echo "Processing $SAMPLE..."
cellranger count \
--id=${SAMPLE} \
--transcriptome=${TRANSCRIPTOME} \
--fastqs=${FASTQ_DIR}/${SAMPLE} \
--sample=${SAMPLE} \
--localcores=16 \
--localmem=128
echo "$SAMPLE completed!"
done
3. 质量检查清单
- 检查 web_summary.html
- 细胞数量合理
- 比对率 > 80%
- 中位基因数 > 500
- 测序饱和度 50-80%
- 线粒体基因比例 < 10%
下一步
完成原始数据处理后,下一步是:
- 质量控制 - 过滤低质量细胞
- 标准化 - 数据归一化
- 降维 - PCA、UMAP
- 聚类 - 识别细胞类型
- 差异分析 - 寻找标志基因
继续学习:模块03:质量控制、聚类与细胞类型注释
参考资源
官方文档
教程
工具
版权声明:本教程为 BioF3 原创学习资源,部分内容组织参考了 scNotebooks 开源项目,并进行了重新编写、本地化和扩展。