Jupyter、交互式分析环境与公共数据库
组学分析通常从两个问题开始:
- 在哪里写下可以重复运行的分析过程?
- 从哪里找到可信的数据,并确认它是否适合自己的问题?
Jupyter Notebook 解决第一个问题,公共数据库解决第二个问题。本章把二者放在一起讲:先学会记录和运行分析,再学会检索、判断和下载公开数据。
学习目标
完成本章后,你应该能够:
- 理解 Notebook 在可重复分析中的作用
- 区分本地 Jupyter、JupyterLab、Google Colab 和服务器环境
- 知道 GEO、SRA、CELLxGENE、HCA、Expression Atlas 分别适合什么数据
- 看懂常见 accession 编号
- 用 AI 辅助检索数据,但保留人工核验
- 建立一个简单的数据探索 Notebook
Notebook 适合做什么
Jupyter Notebook 是一种把代码、说明文字、图表和运行结果放在一起的交互式文档。它适合探索性分析和教学演示。
适合:
- 记录分析思路
- 逐步运行代码
- 快速查看表格和图
- 写下参数解释
- 分享小型分析示例
不适合:
- 长时间无人值守的大流程
- 大规模批处理任务
- 保存敏感数据输出
- 没有版本控制的正式生产流程
推荐做法是:Notebook 用来探索和记录,稳定后把关键步骤整理成 .R、.py 或工作流脚本。
常见交互式环境
Jupyter Notebook
经典 Notebook 界面,适合初学者。文件通常是 .ipynb,内部保存代码单元、Markdown 文本、输出结果和元数据。
启动方式:
jupyter notebook
JupyterLab
JupyterLab 是更完整的工作界面,支持 Notebook、终端、文本编辑器和文件浏览器。做真实项目时更推荐 JupyterLab。
启动方式:
jupyter lab
Google Colab
Google Colab 是云端 Notebook 环境,打开浏览器即可运行 Python。它适合教学和轻量实验,不适合长期保存重要环境或处理隐私数据。
适合:
- 快速试代码
- 课堂演示
- 共享小型 Notebook
- 临时使用 GPU
注意:
- 运行环境可能变化
- 会话可能断开
- 文件需要显式保存
- 不要上传未脱敏的人类样本数据
服务器 Notebook
在真实组学项目中,数据常放在服务器上。可以在服务器启动 JupyterLab,再通过浏览器访问。
一般流程:
ssh aliyun
cd /path/to/project
jupyter lab --no-browser --port 8888
如果需要从本地浏览器访问远程 Notebook,通常会用 SSH 端口转发:
ssh -L 8888:localhost:8888 aliyun
服务器环境要特别注意权限、数据位置和端口安全。
Notebook 的基本结构
Markdown 单元
用来写说明、记录参数和解释结果。
## 质量控制
本步骤过滤低质量细胞:
- `nFeature_RNA < 200`
- `percent.mt > 20`
Code 单元
用来运行代码。
import pandas as pd
metadata = pd.read_csv("data/metadata.csv")
metadata.head()
输出结果
输出可以是表格、图像、日志或错误信息。正式分析时,不建议只依赖 Notebook 里的输出;重要结果应该保存到 results/。
metadata.to_csv("results/metadata_checked.csv", index=False)
Notebook 可重复性清单
一个可维护的 Notebook 至少应该写清楚:
- 数据来源和 accession 编号
- 输入文件路径
- 软件包版本
- 关键参数
- 随机种子
- 输出文件位置
- 每张图对应的代码
- 运行日期
Python 中可以记录版本:
import sys
import pandas as pd
print(sys.version)
print(pd.__version__)
R 中可以记录环境:
sessionInfo()
公共数据库怎么选
不同数据库解决不同问题。先判断你需要的是原始测序数据、处理后的表达矩阵,还是可交互浏览的注释数据。
| 需求 | 优先看哪里 |
|---|---|
| 找论文配套表达矩阵 | GEO |
| 下载原始 FASTQ | SRA / ENA |
| 浏览单细胞注释数据 | CELLxGENE |
| 找人类细胞图谱项目数据 | HCA Data Portal |
| 查基因在组织/细胞中的表达 | Expression Atlas |
常见 accession 编号��
公开数据通常通过 accession 编号追踪。
| 编号 | 常见含义 | 示例 |
|---|---|---|
| GSE | GEO Series,一个研究或数据集 | GSE12345 |
| GSM | GEO Sample,一个样本 | GSM123456 |
| SRP | SRA Study | SRP123456 |
| SRS | SRA Sample | SRS123456 |
| SRX | SRA Experiment | SRX123456 |
| SRR | SRA Run,常用于下载 reads | SRR1234567 |
分析前要确认编号层级。很多新手拿到 GSE 后直接找 FASTQ,会发现真正下载 reads 需要进一步找到对应的 SRR。
主要数据库
GEO
GEO 是 NCBI 维护的功能基因组学数据库,常见于论文数据提交。它可以包含表达矩阵、样本信息、平台信息和补充文件。
网址:https://www.ncbi.nlm.nih.gov/geo/
适合:
- 查找论文配套数据
- 下载处理后的表达矩阵
- 查看样本分组和元数据
- 追踪到 SRA 原始数据
检索建议:
关键词 + 物种 + 技术 + 组织/疾病
例如:
single cell RNA-seq human liver fibrosis
SRA
SRA 是 NCBI 的原始测序数据归档库。需要 FASTQ 时通常会用到它。
网址:https://www.ncbi.nlm.nih.gov/sra/
下载常用 SRA Toolkit:
conda install -c bioconda sra-tools
prefetch SRR1234567
fasterq-dump SRR1234567 --split-files -O data/raw/
注意:
- FASTQ 文件可能很大
- 下载前确认磁盘空间
- 批量下载前先测试一个 run
- 记录 SRR 列表和下载日期
CELLxGENE
CELLxGENE Discover 提供许多可浏览的单细胞数据集,通常可以在线查看 UMAP、细胞类型和基因表达。
网址:https://cellxgene.cziscience.com/
适合:
- 快速浏览单细胞数据
- 查看细胞类型注释
- 寻找可下载的 h5ad 数据
- 比较公开数据中的基因表达模式
使用建议:
- 先在线检查数据是否符合研究问题
- 下载前确认样本、组织、物种和处理流程
- 注意数据是否已经标准化或整合
Human Cell Atlas
Human Cell Atlas 关注人类细胞参考图谱。HCA Data Portal 提供社区生成的多组学开放数据。
网址:https://data.humancellatlas.org/
适合:
- 查找人类组织和器官图谱
- 获取大型参考数据
- 了解细胞类型注释和 atlas 项目
- 作为数据整合和注释参考
Expression Atlas / Single Cell Expression Atlas
EMBL-EBI 的 Expression Atlas 和 Single Cell Expression Atlas 提供基因表达查询和单细胞表达浏览。
适合:
- 查询某个基因在不同组织或细胞中的表达
- 浏览经过整理的表达数据
- 做初步假设生成
- 辅助解释 marker gene
AI 辅助数据检索
AI 很适合帮助你缩小搜索范围,但不能替代数据库原始页面。
适合让 AI 做:
- 根据研究问题生成检索关键词
- 解释 accession 编号层级
- 总结某个数据集页面的字段含义
- 根据样本表检查分组是否清楚
- 生成下载脚本草稿
不适合让 AI 做:
- 直接断言某个数据集一定适合你的研究
- 编造不存在的 accession 编号
- 代替你检查原始数据库页面
- 忽略伦理、隐私和数据使用限制
推荐提示词:
我想找人类肝纤维化的单细胞 RNA-seq 公开数据。请帮我生成 GEO、SRA 和 CELLxGENE 的检索关键词,并说明每个关键词适合查什么。不要编造 accession 编号。
找到候选数据后,再让 AI 做核对清单:
下面是一个 GEO 数据集页面的信息。请帮我提取物种、组织、样本数、处理组、测序平台、是否有原始数据、是否有表达矩阵。无法确定的地方请标为“需要人工确认”。
实践练习:建立第一个数据探索 Notebook
下面用模拟表达矩阵演示 Notebook 的基本结构。
1. 准备数据
import numpy as np
import pandas as pd
np.random.seed(42)
n_genes = 100
n_cells = 50
expression_matrix = np.random.poisson(lam=5, size=(n_genes, n_cells))
gene_names = [f"Gene_{i+1}" for i in range(n_genes)]
cell_names = [f"Cell_{i+1}" for i in range(n_cells)]
expr = pd.DataFrame(
expression_matrix,
index=gene_names,
columns=cell_names,
)
expr.head()
2. 计算基础指标
cell_total_counts = expr.sum(axis=0)
gene_mean_expression = expr.mean(axis=1)
qc = pd.DataFrame({
"cell": cell_total_counts.index,
"total_counts": cell_total_counts.values,
})
qc.head()
3. 绘制质量控制图
import matplotlib.pyplot as plt
fig, axes = plt.subplots(1, 2, figsize=(10, 4))
axes[0].hist(cell_total_counts, bins=20, color="#3B82F6", edgecolor="white")
axes[0].set_xlabel("Total counts per cell")
axes[0].set_ylabel("Number of cells")
axes[1].hist(gene_mean_expression, bins=20, color="#10B981", edgecolor="white")
axes[1].set_xlabel("Mean expression per gene")
axes[1].set_ylabel("Number of genes")
plt.tight_layout()
plt.show()
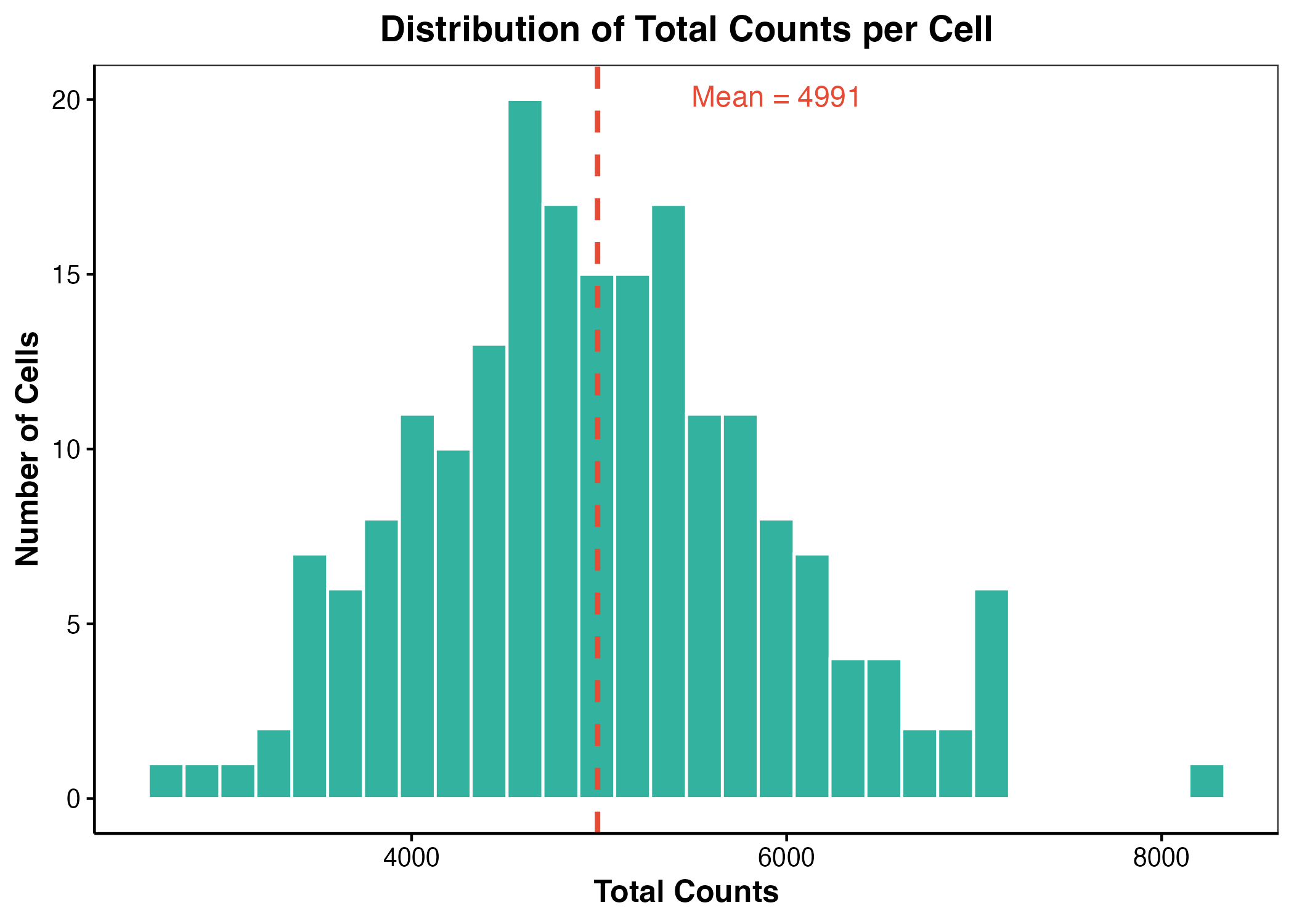
图 1:细胞总表达量分布示例。真实项目中可以用它检查测序深度和低质量细胞。
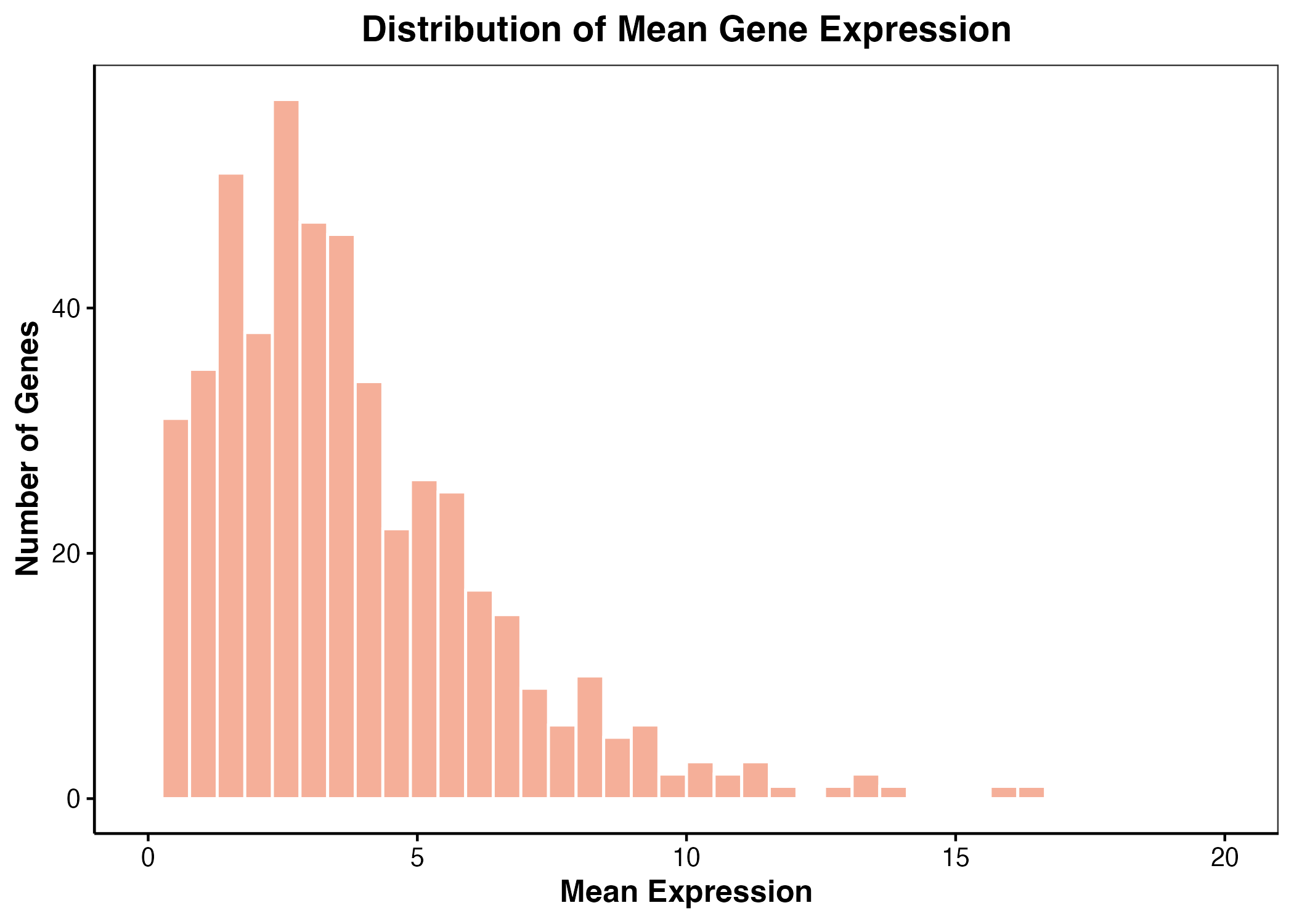
图 2:基因平均表达量分布示例。真实项目中可以用它观察高表达和低表达基因。
4. 保存结果
from pathlib import Path
Path("results/tables").mkdir(parents=True, exist_ok=True)
qc.to_csv("results/tables/qc_summary.csv", index=False)
数据下载前检查清单
- 数据是否来自可信数据库
- accession 编号是否正确
- 物种、组织、疾病和技术是否符合问题
- 样本数是否足够
- 是否有清楚的 metadata
- 是否有处理后的表达矩阵
- 是否需要下载原始 FASTQ
- 是否有数据使用限制
- 是否有对应论文或预印本
- 是否记录了下载日期和数据库链接
下一步
完成本章后,建议继续学习:
参考资源
- Jupyter 官方文档:https://docs.jupyter.org/
- JupyterLab 文档:https://jupyterlab.readthedocs.io/
- Google Colab:https://colab.research.google.com/
- GEO:https://www.ncbi.nlm.nih.gov/geo/
- SRA:https://www.ncbi.nlm.nih.gov/sra/
- CELLxGENE:https://cellxgene.cziscience.com/
- Human Cell Atlas Data Portal:https://data.humancellatlas.org/
- Expression Atlas:https://www.ebi.ac.uk/gxa/