08 TCR/BCR 测序分析
08 TCR/BCR 测序分析
T 细胞受体(TCR)和 B 细胞受体(BCR)单细胞测序把免疫学里最核心的问题——"同一个细胞是哪个克隆"——放到了分辨率最高的层面。10x Genomics 的 V(D)J 试剂盒能同时测出一个细胞的 GEX(基因表达)和 V(D)J(受体序列),后续分析的主线就是:把 V(D)J 结果和 GEX 对象绑起来,再在每个聚类里看克隆是否扩增、扩增来自哪些通路。
本节介绍两种最常见的做法:用 Seurat 把 clonotype 作为 metadata 加进对象(适合快速可视化),以及用 scRepertoire 做更完整的克隆分析(适合多样本比较)。
这种数据能回答的核心问题
V(D)J 测序解决一个 RNA-seq 不能回答的问题:两个 T 细胞看上去几乎一样(同样的 CD3+CD4+ memory marker),但它们识别的抗原可能完全不同。
具体场景:
- 抗原识别追踪:肿瘤里的 T 细胞中,哪些是真在打肿瘤、哪些是路过的旁观者?看克隆扩增 — 扩增的更可能是抗原特异性
- 疫苗 / 治疗响应:免疫治疗后,新出现的扩增克隆有哪些?克隆是从哪个亚群里冒出来的?
- 克隆与功能状态绑定:同一个克隆的细胞是分散在 effector / memory / 耗竭多个状态,还是集中在某一个?这告诉你这个克隆的命运
适合做 V(D)J 的项目:肿瘤免疫、感染响应、自免、疫苗。不适合:发育图谱、稳态健康成人 PBMC(克隆多样性高、扩增稀少,跑了也没什么发现)。
V(D)J 数据的基本概念
| 术语 | 说明 |
|---|---|
| V / D / J | 可变区的三段基因片段,重组组合决定了受体多样性 |
| CDR3 | 受体上最变异的区域,是克隆身份的"指纹" |
| Clonotype | 基于 CDR3 序列等定义的一组相同来源细胞 |
| Clonal expansion | 某个 clonotype 在样本中出现多次(抗原驱动) |
TCR 由 α 链和 β 链组成,通常 β 链更能代表克隆身份;BCR 由重链和轻链组成,重链负责识别。
Cell Ranger 的 VDJ 流程
GEX 和 VDJ 来自同一样本的不同文库。Cell Ranger 的 VDJ 子命令会输出 contigs、clonotypes 和每个细胞的注释:
cellranger vdj \
--id=sample_tcr \
--reference=refdata-cellranger-vdj-GRCh38-alts-ensembl-7.0.0 \
--fastqs=/path/to/fastqs \
--sample=sample_name
运行完会得到 outs/filtered_contig_annotations.csv,每行是一个 contig,带 barcode、chain(TRA/TRB)、v/d/j_gene、CDR3 序列和 raw_clonotype_id。
用 Seurat 把 clonotype 当 metadata 加进来
这是最简单的做法:把 VDJ 表聚合到 barcode 级,作为 metadata 加到 GEX 的 Seurat 对象上,就能在 UMAP 上按 clonotype 染色。
library(Seurat)
library(dplyr)
# 先有一个已分析好的 GEX Seurat 对象(03 / 04 章的产物)
gex <- readRDS("pbmc_gex.rds")
# 读取 Cell Ranger VDJ 的 contig annotation
tcr <- read.csv("filtered_contig_annotations.csv")
# 把每个 barcode 聚合成一行,保留 clonotype 和 CDR3
clonotypes <- tcr %>%
group_by(barcode) %>%
summarise(
clonotype_id = first(raw_clonotype_id),
cdr3 = paste(unique(cdr3), collapse = ";")
) %>%
tibble::column_to_rownames("barcode")
# 加到 Seurat metadata(只会加到匹配得上 barcode 的细胞)
gex <- AddMetaData(gex, metadata = clonotypes)
# 在 UMAP 上看前 10 个最大的 clonotype
top_clones <- head(sort(table(gex$clonotype_id), decreasing = TRUE), 10)
gex$top_clone <- ifelse(gex$clonotype_id %in% names(top_clones),
gex$clonotype_id, NA)
DimPlot(gex, group.by = "top_clone", order = TRUE)
这样就能直观看到"扩增最多的克隆落在哪些聚类"。但这种做法只是展示,还不能做克隆多样性、克隆追踪之类的定量分析。
用 scRepertoire 做完整克隆分析
scRepertoire 把常见克隆分析打包成一套函数,多样本场景下尤其方便:
library(scRepertoire)
# 载入多个样本的 VDJ 结果
tcr_list <- list(
sample1 = read.csv("sample1/filtered_contig_annotations.csv"),
sample2 = read.csv("sample2/filtered_contig_annotations.csv")
)
# 合并并标注样本来源
combined <- combineTCR(
tcr_list,
samples = c("S1", "S2"),
ID = c("P1", "P1")
)
# 克隆多样性(样本间对比)
clonalDiversity(combined, cloneCall = "gene")
# 克隆稳态:按克隆占比划档看分布
clonalHomeostasis(combined, cloneCall = "gene")
# 把 repertoire 信息合并进 Seurat GEX 对象
gex <- combineExpression(combined, gex,
cloneCall = "gene",
proportion = TRUE)
combineExpression 会在 gex@meta.data 里新增 cloneType 等字段,标记每个细胞属于"单拷贝/小克隆/中克隆/大克隆"。再配合 DimPlot(gex, group.by = "cloneType") 就能看到克隆扩增在 UMAP 空间的分布。
BCR 的流程几乎完全一致,只是用 combineBCR 代替 combineTCR。
真实示例:8 个样本跑一遍 scRepertoire
配套脚本 module09_tcr_sci.R 用 scRepertoire 包自带的 8 个 10x VDJ 样本(4 个病人 P17~P20 各自 B/L 两个部位,contig_list)和配套的 500 细胞 Seurat 对象(scRep_example)演示完整流程。包里的数据是真实样本的 filtered_contig_annotations,不需要下载。
Rscript scripts/single-cell/sc09_tcr_sci.R
脚本的顺序是:combineTCR 合并 8 个样本 → 算各样本独立克隆数、丰度分布、稳态、Shannon 多样性、样本两两之间的 Morisita 重叠 → combineExpression 把克隆信息写进 Seurat 的 metadata,在 UMAP 上按克隆大小上色。
每张图看什么
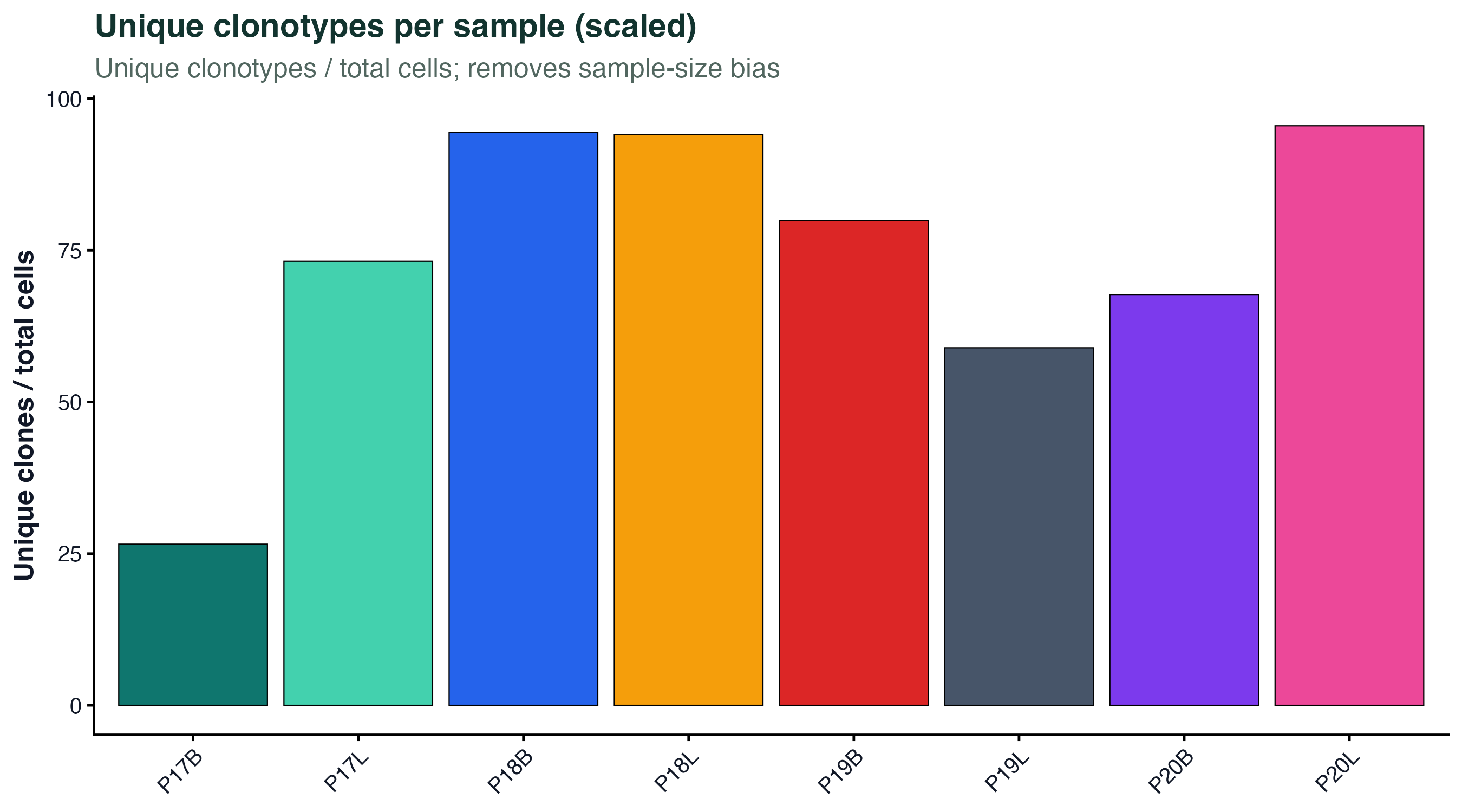
图 1:每个样本的独特克隆数除以该样本的 T 细胞总数。scale=TRUE 去掉样本大小差异,数值越低说明克隆集中度越高。可以直接看到 P19L、P20B 这种组织/淋巴样本比外周血样本更"少而大"。
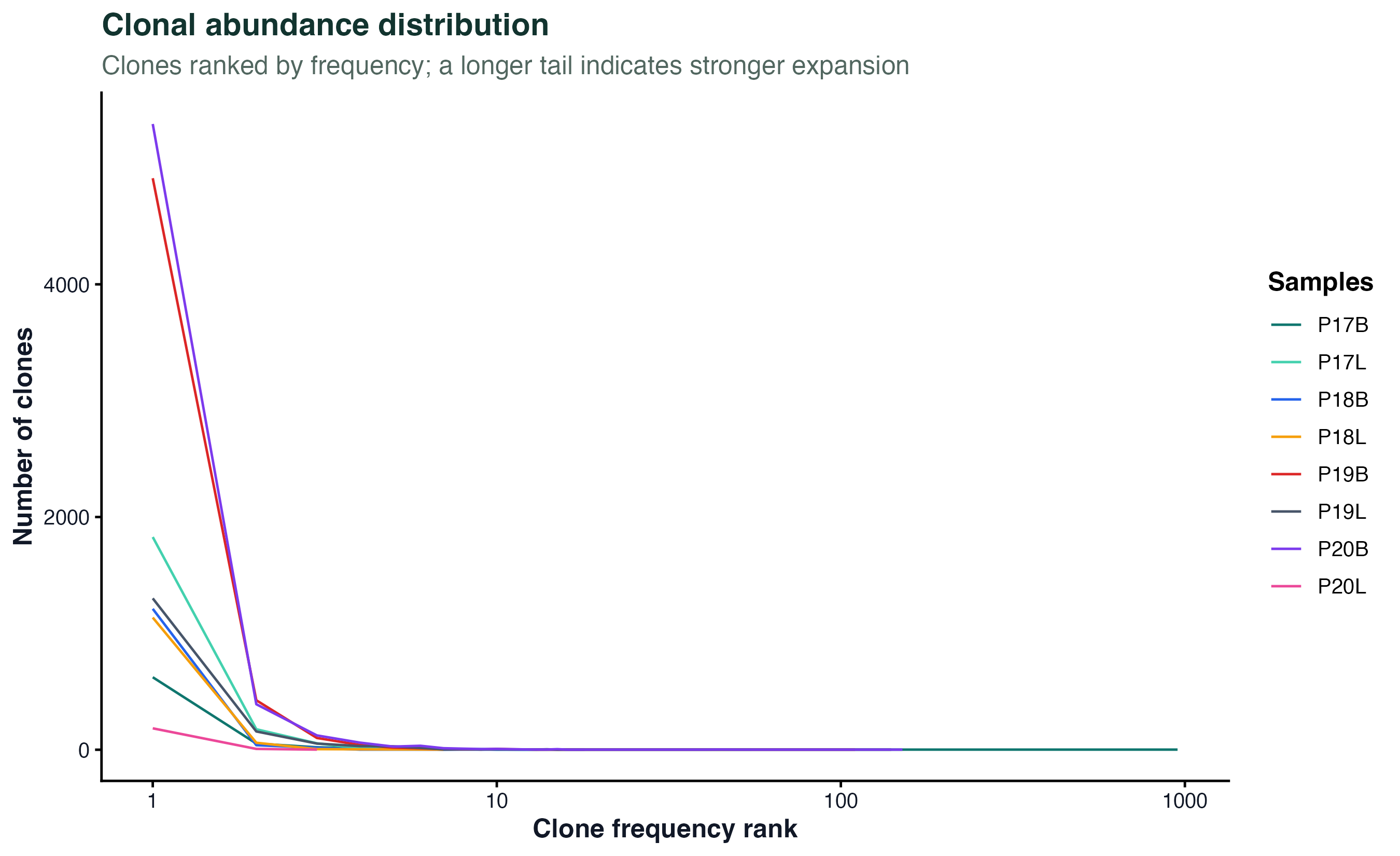
图 2:克隆按频率排序,纵轴是该频率上的克隆数。曲线长尾越平、越往右延伸说明"有少数克隆扩增得特别大";曲线集中在左下角说明所有克隆都很小、差异不明显。
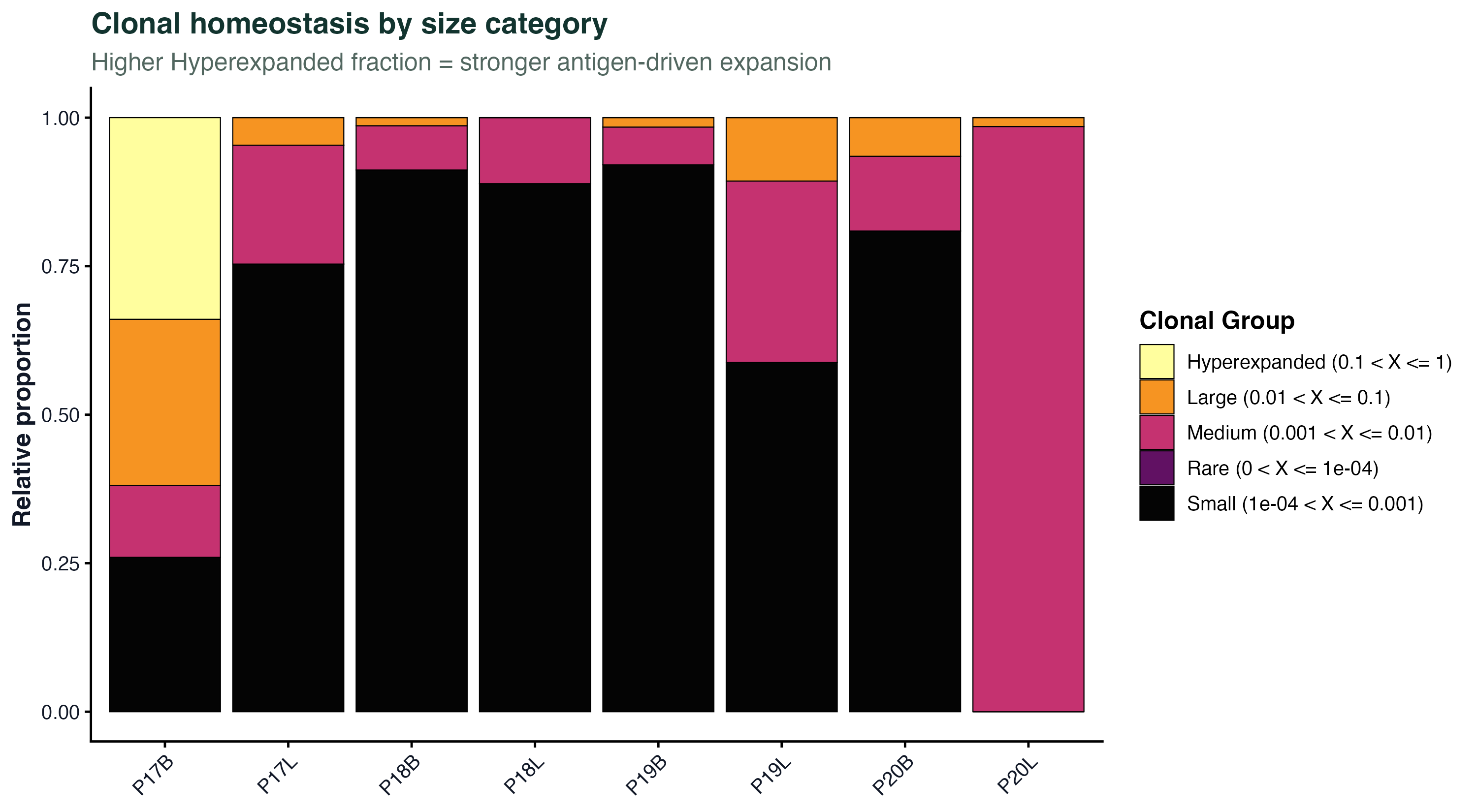
图 3:把每个样本的克隆按大小分成 Rare / Small / Medium / Large / Hyperexpanded 五档,画堆叠条形。Hyperexpanded 占比越高说明样本里有显著抗原驱动扩增。这是比较治疗前后、组织间免疫反应强度最直观的一张图。
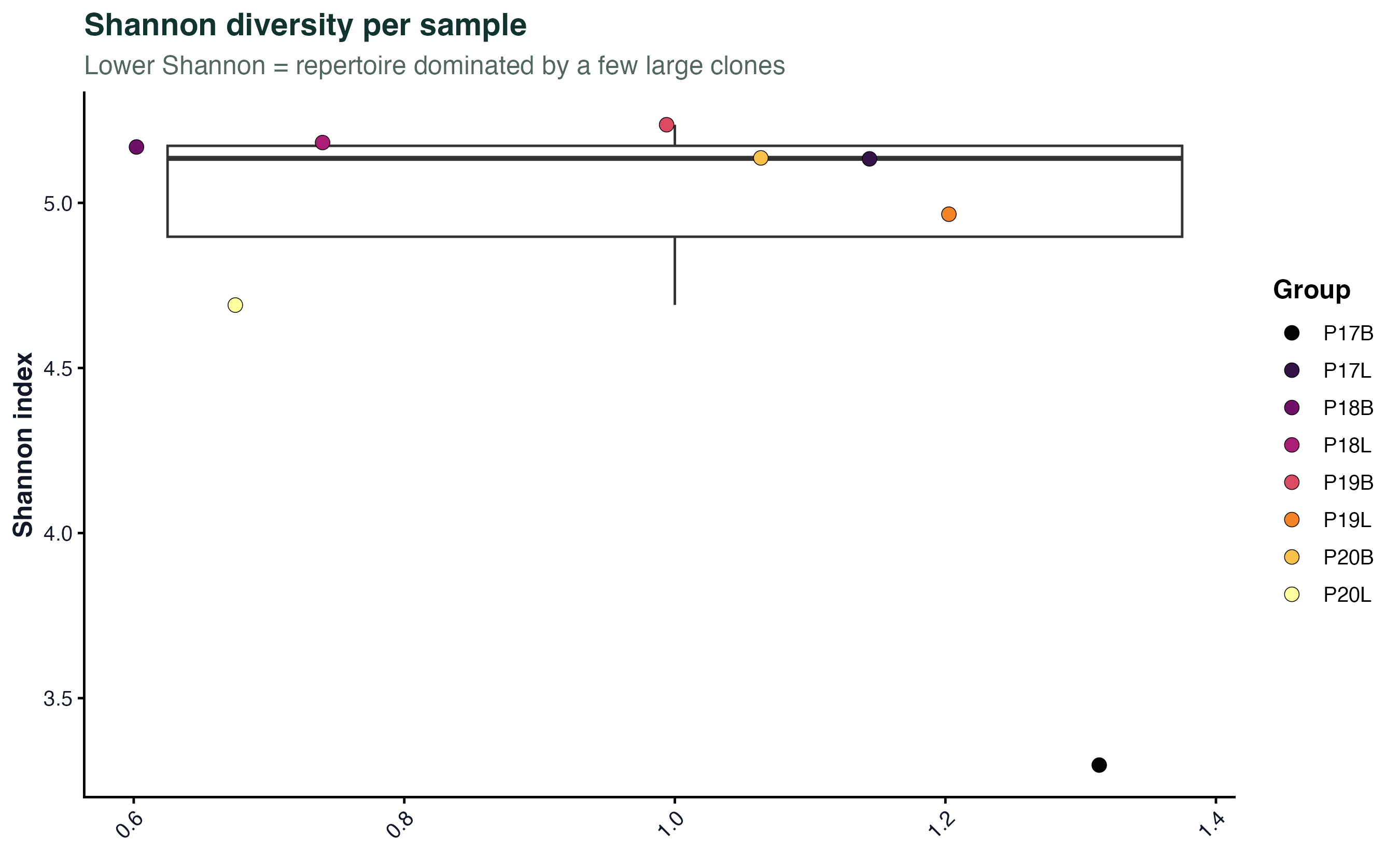
图 4:每个样本的 Shannon 多样性指数。Shannon 越低代表克隆被少数几个大克隆主导。真实项目里,除了 Shannon,还会一起画 inv.simpson、chao1,但 scRepertoire 2.x 的 clonalDiversity 一次只画一个指标,要多画几张。
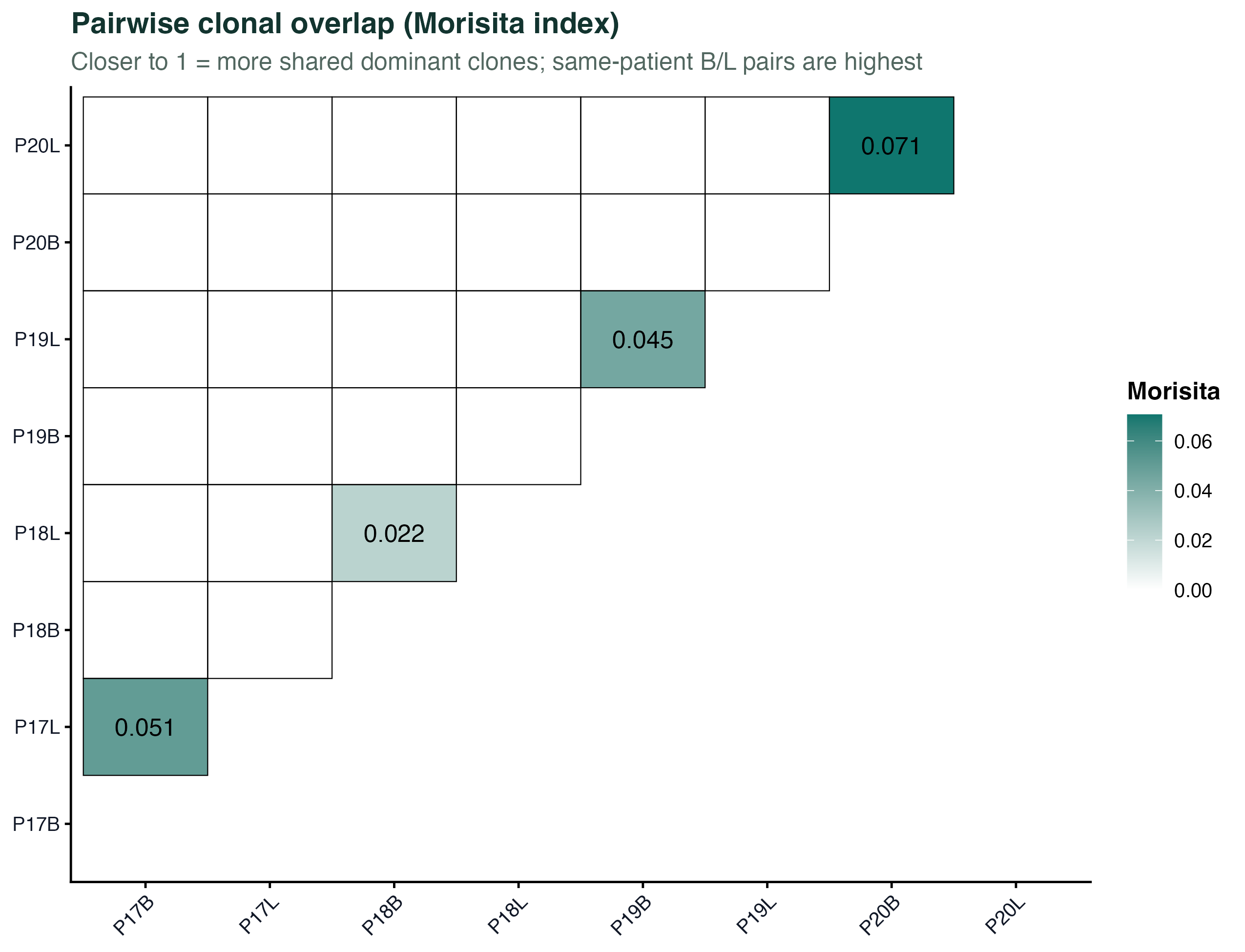
图 5:样本两两之间用 Morisita 指数衡量共享克隆的热图。越接近 1 说明两个样本里大小靠前的克隆越一致。可以看到同一个病人不同部位(P17B-P17L、P19B-P19L)之间的共享明显高于跨病人。
图 6:把上面算出的 cloneSize 档位写到 Seurat 的 metadata 里,直接在 UMAP 上按档位染色。每个点是一个 T 细胞,灰色=几乎没扩增,红/紫色=超大克隆。连 GEX 聚类一起看,就能回答"哪些亚群里扎堆出现了扩增克隆"。
套到自己数据上
脚本里的 combineTCR 直接换成自己 read.csv("filtered_contig_annotations.csv") 结果的 list,samples 和 tissue_ids 换成自己的样本/部位标签即可。scRep_example 那步换成自己已经走过 03~04 章标准流程的 Seurat 对象;combineExpression 的 cloneSize 档位按样本规模调,样本很小时把最大档降到 0.05 以下能避免上不了色。BCR 的流程几乎完全一样,把 combineTCR 换成 combineBCR 即可。
常见分析方向
- 抗原特异性定位:拿扩增 top 克隆的 CDR3,和 VDJdb、IEDB 等公共数据库里已知抗原特异性的 CDR3 比对,推测它们识别什么。
- 克隆与细胞状态关联:看扩增 clonotype 落在哪些聚类(耗竭 T?效应 T?),结合差异基因解释功能状态。
- 治疗前后追踪:配对样本里同一个克隆在前后的频率、亚群归属变化,是免疫治疗响应研究的主线。
常见坑
坑 1:GEX 和 VDJ 的 barcode 来自不同 library
10x V(D)J 试剂盒里 GEX 和 TCR / BCR 是两份独立的 library(不同 sequencing run)。barcode 是同一套,但 fastq 是分开下机的。读 contig csv 后 AddMetaData 时只匹配 barcode 重叠那部分细胞 — 没有 TCR 信息的细胞 metadata 列是 NA,这是正常的。
坑 2:把所有 contig 都当独立克隆
一个 T 细胞可能有 2 条 α + 1 条 β(双 TCR)或 1 条 α + 1 条 β。直接用 contig 数当克隆数会错,要用 raw_clonotype_id 这个汇总后的 ID。scRepertoire::combineTCR 会处理双链问题,比手动聚合可靠。
坑 3:稀有克隆的统计意义
样本里出现 1 次的克隆叫 singleton — 不能说"这个克隆没扩增"。它可能是真的稀有,也可能是这个克隆的其他成员没被采到。用 clonalDiversity 看整体多样性,而不是去解读单个 singleton。
坑 4:跨样本比较不做样本大小校正
P19L 5000 个细胞 vs P17B 500 个细胞,直接比克隆数 P19L 一定多。要用 clonalQuant(scale = TRUE) 或归一化到每千细胞克隆数后比较,否则结论是"细胞多的样本克隆多"这种空话。
坑 5:BCR 数据不做 hypermutation 分析就当 TCR 用
BCR 在抗原刺激下会发生 somatic hypermutation,同一克隆的不同细胞 CDR3 序列也会有差异。如果用 TCR 那套"完全相同 CDR3 = 同克隆"判定,BCR 会把同一克隆的不同子代算成不同克隆。BCR 项目用 combineBCR + scoper 做克隆型聚类,不要直接用 raw_clonotype_id。
下载资源
下一步
接着深入:
- 09 空间转录组学 — 把克隆信息映射到组织切片上,能看到扩增克隆是否聚集在 TLS(三级淋巴结构)这类局部位点
- 03 质量控制、聚类与细胞类型注释 — V(D)J 信息只在 GEX 已经注释好的对象上才有意义,先做完 GEX 标准流水线
横向延伸:
- VDJdb 抗原-CDR3 数据库 — 把 top 扩增克隆的 CDR3 序列粘进去,看是否匹配已知抗原
- scRepertoire 教程 — 完整的克隆分析方法论