02 DEP 差异蛋白分析
02 DEP 差异蛋白分析
DEP(Differential Enrichment analysis of Proteomics data)把蛋白组差异分析的完整流程封装成一条管线:过滤 → 归一化 → 插补 → limma 差异检验 → 多重校正。底层用的是 limma,所以统计学上和 bulk RNA-seq 的差异分析是同一套框架。
本章用 DEP 自带的 UbiLength 数据走一遍完整流程。数据是 HeLa 细胞在不同泛素链长度处理下的蛋白组(4 个条件 × 3 个重复 = 12 个样本,约 3000 个蛋白)。
完整流程
library(DEP)
data(UbiLength)
data(UbiLength_ExpDesign)
# 1. 确保蛋白名唯一
data_unique <- make_unique(UbiLength, "Gene.names", "Protein.IDs", delim = ";")
# 2. 构建 SummarizedExperiment
lfq_cols <- grep("LFQ.intensity.", colnames(data_unique))
data_se <- make_se(data_unique, lfq_cols, UbiLength_ExpDesign)
# 3. 过滤:每组至少 2 个样本有值
data_filt <- filter_missval(data_se, thr = 0)
# 4. VSN 归一化
data_norm <- normalize_vsn(data_filt)
# 5. 缺失值插补(MinProb:从左尾采样)
data_imp <- impute(data_norm, fun = "MinProb", q = 0.01)
# 6. limma 差异检验
data_diff <- test_diff(data_imp, type = "control", control = "Ctrl")
# 7. 标记显著蛋白
dep <- add_rejections(data_diff, alpha = 0.05, lfc = 1)
test_diff(type = "control", control = "Ctrl") 会自动生成所有"XX vs Ctrl"的对比。如果要做两两比较,用 type = "all"。
真实示例:UbiLength 上的 DEP 分析
配套脚本 prot02_dep_sci.R 把上面的流程完整跑了一遍,输出 6 张图:
Rscript scripts/proteomics/prot02_dep_sci.R
图 1:缺失值热图
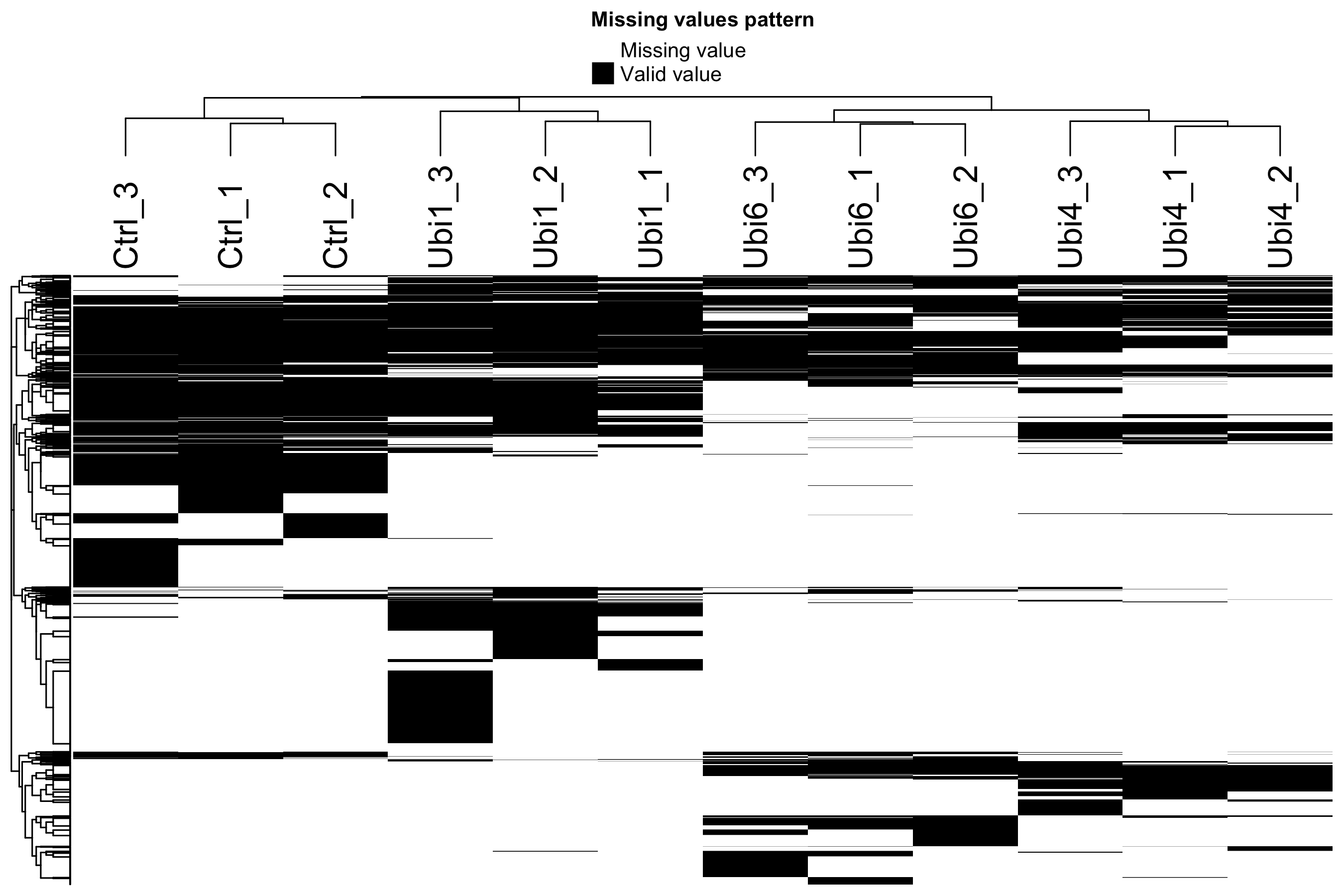
每行是一个蛋白,每列是一个样本。黑色 = 有值,灰色 = 缺失。蛋白组的缺失不是随机的 —— 低丰度蛋白在所有样本里都容易缺失(整行灰色),这就是 MNAR 的典型模式。
这张图在 QC 阶段最重要的用途:看有没有某个样本缺失率异常高(整列灰色比别的多很多)。如果有,要考虑是不是该样本的质谱跑坏了。
图 2:归一化前后的强度分布
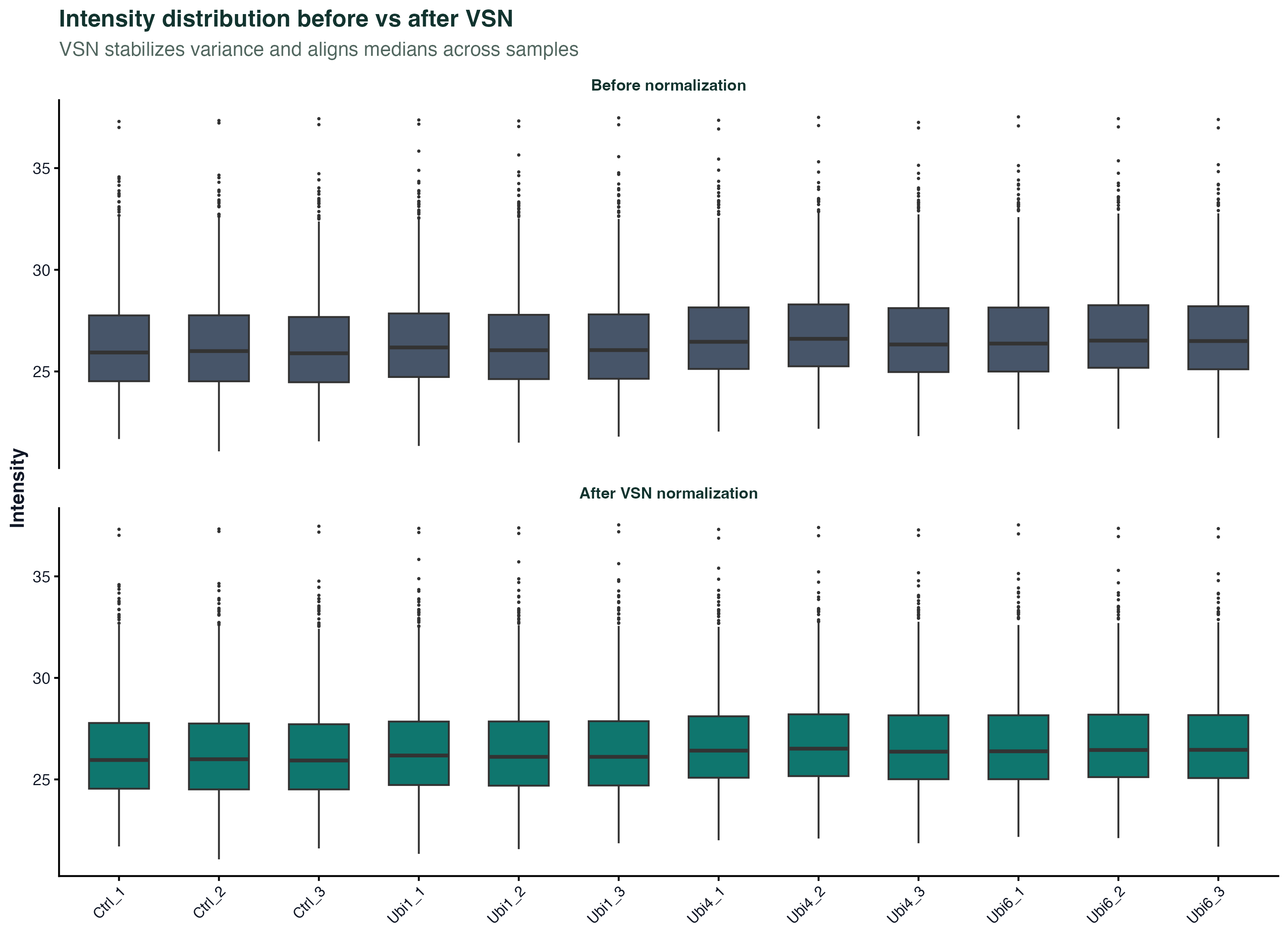
上面是归一化前,下面是 VSN 归一化后。归一化的目标是让所有样本的中位线对齐、方差稳定。如果归一化前某些样本的 box 明显偏高或偏低,说明上样量或质谱响应有系统偏差。
VSN(Variance Stabilizing Normalization)和 bulk RNA-seq 里的 vst 思路一样:让高强度和低强度蛋白的方差都差不多,后续 limma 的 t 检验才不会被少数高丰度蛋白主导。
图 3:PCA
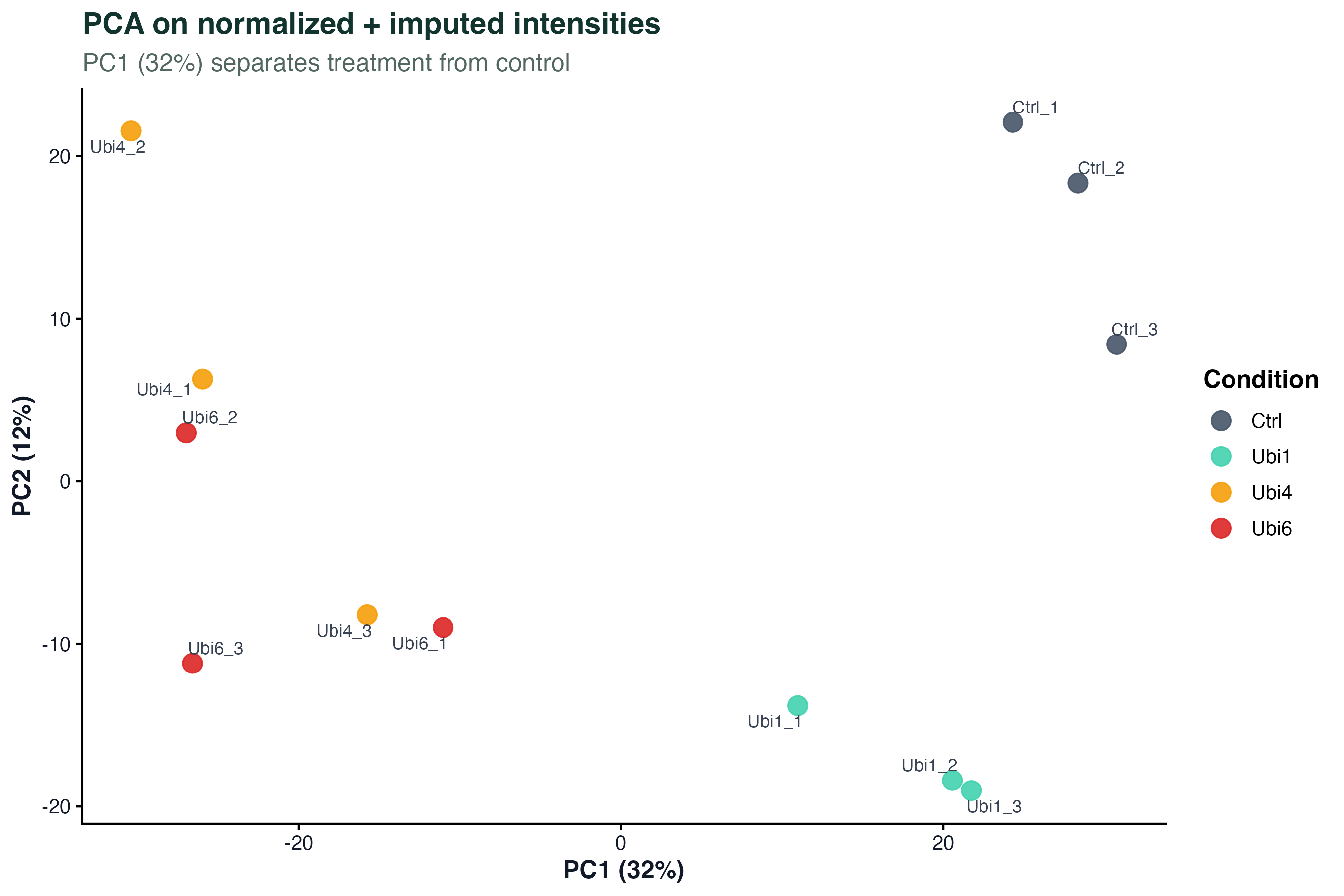
归一化 + 插补之后做 PCA。颜色是条件(Ctrl / Ubi1 / Ubi4 / Ubi6)。PC1 应该把处理组和对照组分开。如果同一条件的重复散得很开,说明技术变异大或者某个重复有问题。
UbiLength 里 Ubi6(最长泛素链)和 Ctrl 分得最远,Ubi1 和 Ctrl 最近 —— 符合生物学预期:泛素链越长,蛋白组变化越大。
图 4:样本相关性热图
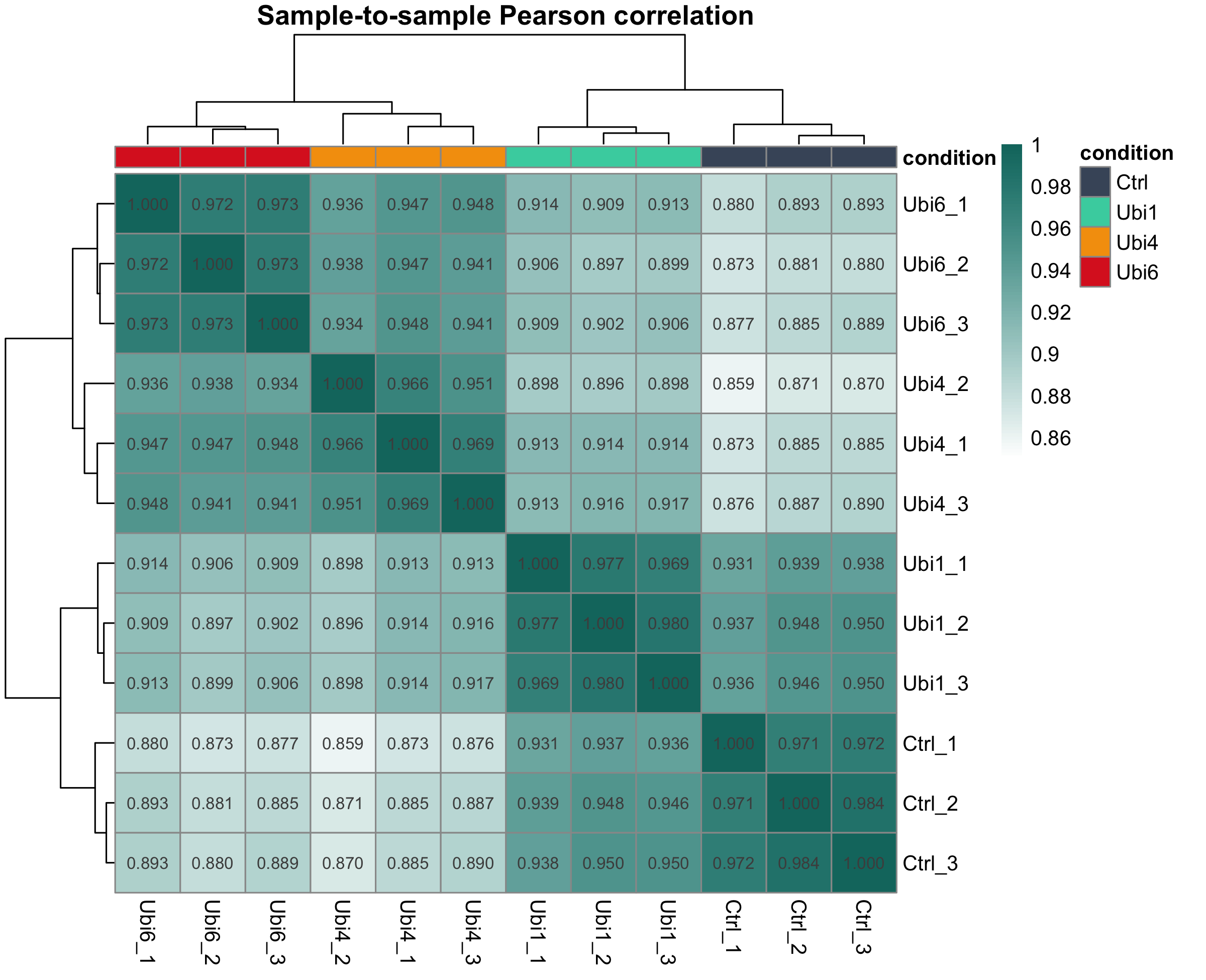
样本两两之间的 Pearson 相关。同一条件的样本之间相关性应该最高(对角线附近的深色块)。如果某个样本和同组的相关性反而低于和别组的,要回头看 QC。
图 5:火山图(Ubi6 vs Ctrl)
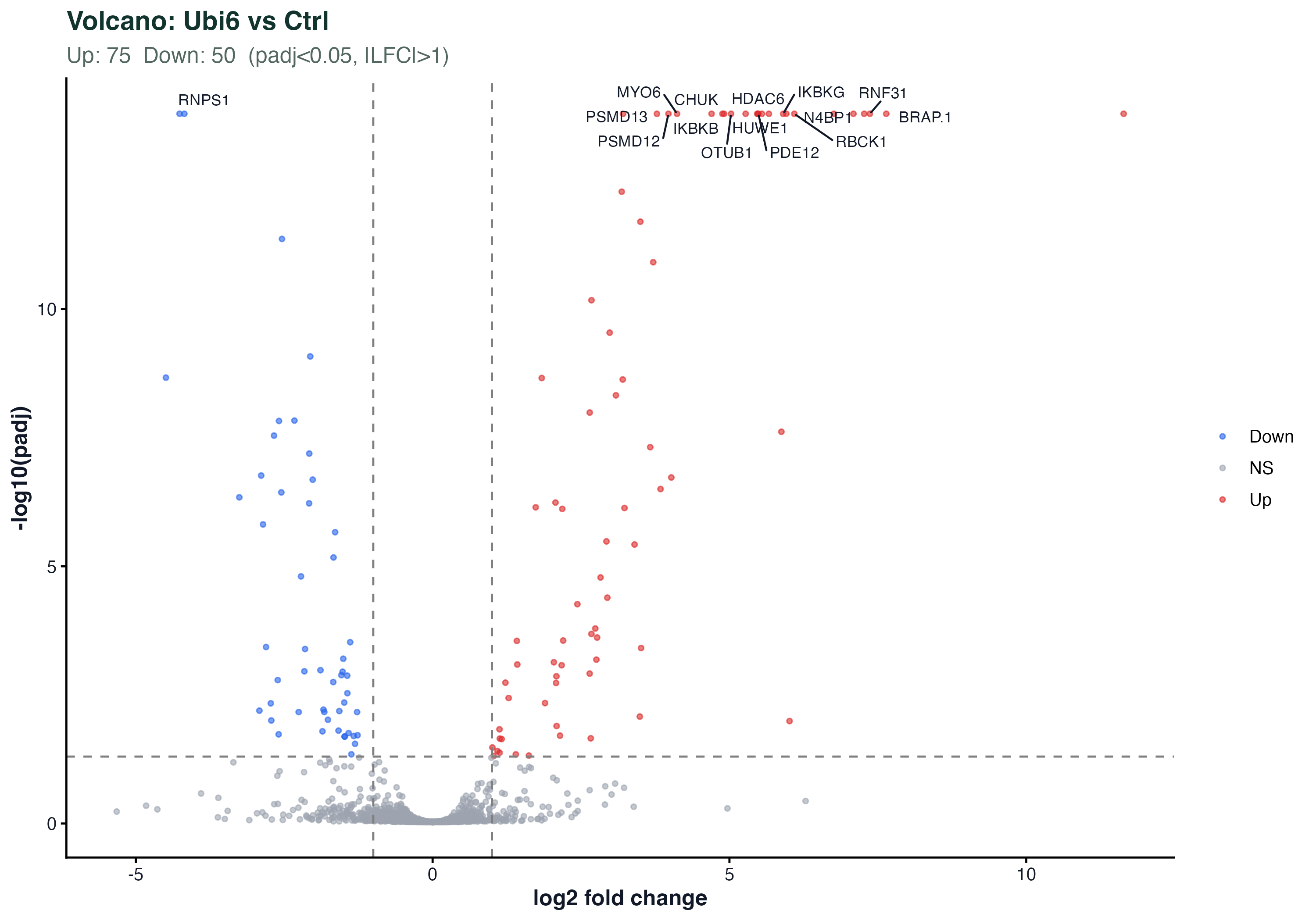
横轴 log2 fold change,纵轴 -log10(padj)。红色 = 上调(Ubi6 里更高),蓝色 = 下调。标注了 padj 最小的 15 个蛋白。
和 RNA-seq 火山图的读法完全一样。蛋白组的特点是:点数少(几千 vs 几万)、效应大小通常比转录组小(蛋白翻译后调控会缓冲 mRNA 变化)。
图 6:每个对比的显著蛋白数
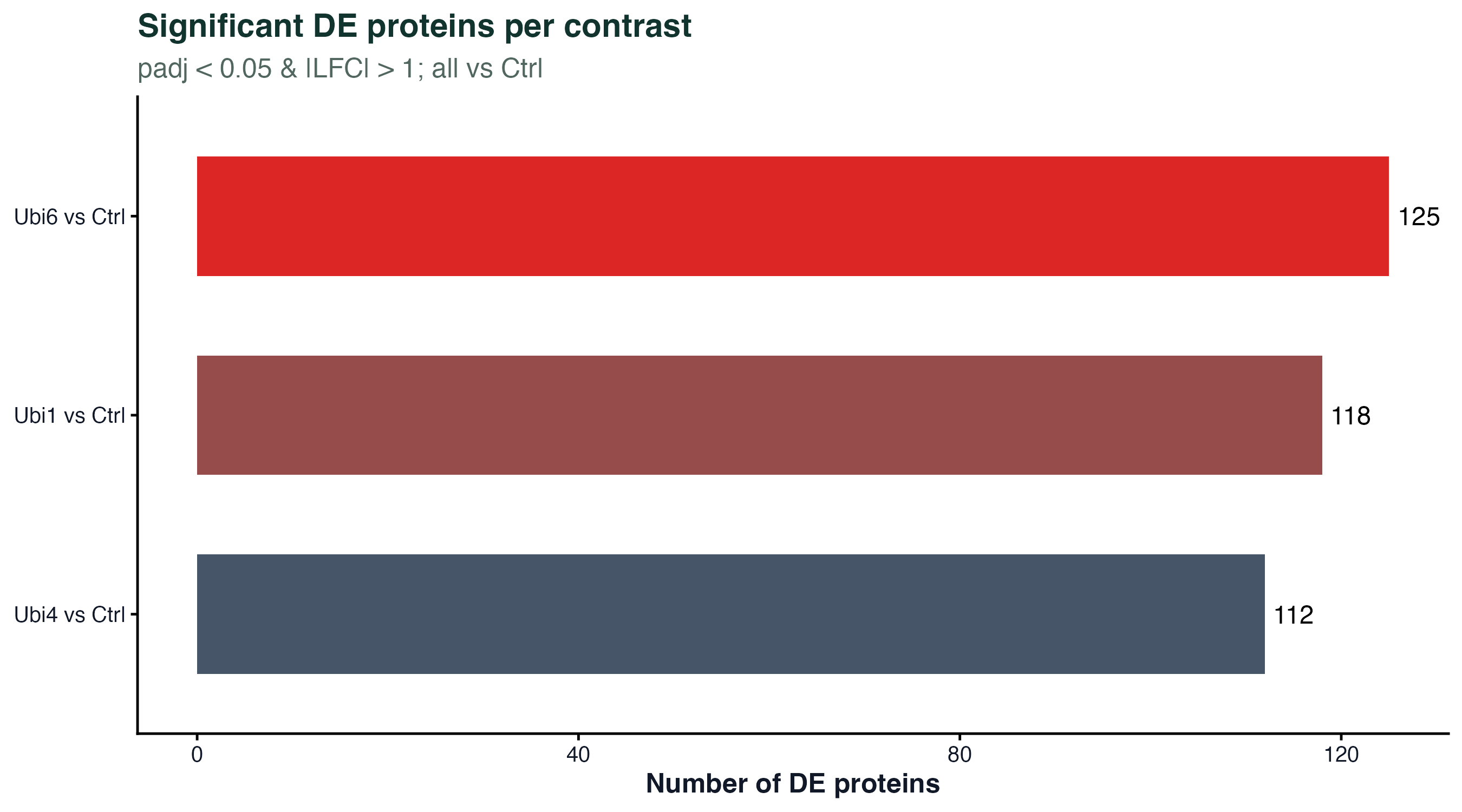
三个对比(Ubi1/Ubi4/Ubi6 vs Ctrl)各自有多少显著差异蛋白。Ubi6 最多、Ubi1 最少,和 PCA 的分离程度一致。
套到自己数据上
脚本的前 10 行把 UbiLength 换成自己的 proteinGroups.txt 就行:
data <- read.delim("proteinGroups.txt")
data <- data[data$Reverse != "+", ]
data <- data[data$Potential.contaminant != "+", ]
data_unique <- make_unique(data, "Gene.names", "Protein.IDs", delim = ";")
UbiLength_ExpDesign 换成自己的样本表(label / condition / replicate 三列)。
几个常见调整:
- DIA 数据:DIA-NN 输出的
report.pg_matrix.tsv已经是宽表,列名就是样本名,直接make_se即可 - 插补策略:如果缺失率 > 50%,MinProb 可能不够好,试
fun = "knn"或fun = "mixed" - 多因子设计:
test_diff(type = "manual", test = ...)可以传自定义 contrast
下载资源
下一步
常见坑
坑 1:插补后忘了检查 PCA 是否合理
插补会人为填充数据,可能引入假信号。插补完应该立刻做 PCA 看样本是否按条件分组。如果插补后 PCA 的分离比插补前更好,要警惕插补方法是否引入了偏差(尤其是 MinProb 在小样本时容易让同组样本人为聚拢)。
坑 2:filter_missval 的阈值设太松
filter_missval(data_se, thr = 0) 意味着只要有一个样本有值就保留。这会保留很多只在 1-2 个样本里测到的蛋白,插补后做差异分析全靠插出来的值。建议 thr = 1(每组至少 2/3 样本有值)或更严格,对不确定的蛋白标注"低置信度"。
坑 3:多个对比共用同一套 p 值阈值
test_diff(type = "all") 同时做所有两两比较,每个对比各自做了 BH 校正。但如果你从多个对比中挑 "任一对比 padj < 0.05" 的蛋白,相当于又做了一层多重检验没校正。建议对最终候选列表再做一次整体 FDR 控制。
坑 4:火山图里把所有对比画在一张图上
三个对比叠在同一张火山图上会让点互相遮挡,无法分辨哪个点属于哪个对比。正确做法是 facet 分面或并排画图,每个对比独立展示。
下一步
接着深入:
- 03 功能富集与 Reactome 通路 — 差异蛋白列表的生物学解读
- 05 缺失值的两种来源与策略对比 — 深入理解插补策略的影响
横向延伸:
- bulk RNA-seq 02 差异表达分析 — limma 在转录组中的应用,和蛋白组统计框架一致
- 多组学整合 02 数据清洗 — 蛋白组作为整合输入的预处理要求