05 MOFA2 因子分析实战
05 MOFA2 因子分析实战
MOFA2(Multi-Omics Factor Analysis v2)用概率矩阵分解从多层组学数据中提取潜因子。每个因子捕捉一种跨组学的共同变异模式,可以理解为"多组学版的 PCA"。本节用 MOFA2 自带的 CLL(慢性淋巴细胞白血病)数据集走一遍完整流程。
数据准备与模型训练
MOFA2 接受长格式数据框或 MultiAssayExperiment 对象。CLL 数据集包含 4 个组学层(mRNA、Methylation、Mutations、Drug response)和 200 个样本。
library(MOFA2)
library(ggplot2)
# 载入 CLL 示例数据
file <- system.file("extdata", "CLL_data.hdf5", package = "MOFA2")
model <- load_model(file)
# 查看模型基本信息
model
如果从头训练模型:
# 从矩阵列表创建 MOFA 对象
data_list <- list(
mRNA = rna_final,
Methylation = meth_final,
Protein = prot_final
)
mofa_obj <- create_mofa(data_list)
# 设置参数
data_opts <- get_default_data_options(mofa_obj)
model_opts <- get_default_model_options(mofa_obj)
model_opts$num_factors <- 15
train_opts <- get_default_training_options(mofa_obj)
train_opts$convergence_mode <- "slow"
train_opts$seed <- 42
mofa_obj <- prepare_mofa(mofa_obj,
data_options = data_opts,
model_options = model_opts,
training_options = train_opts)
# 训练(需要 Python 环境中安装 mofapy2)
model <- run_mofa(mofa_obj, outfile = "mofa_model.hdf5")
方差解释
第一步是看每个因子在各组学层解释了多少方差。这决定了哪些因子值得深入分析。
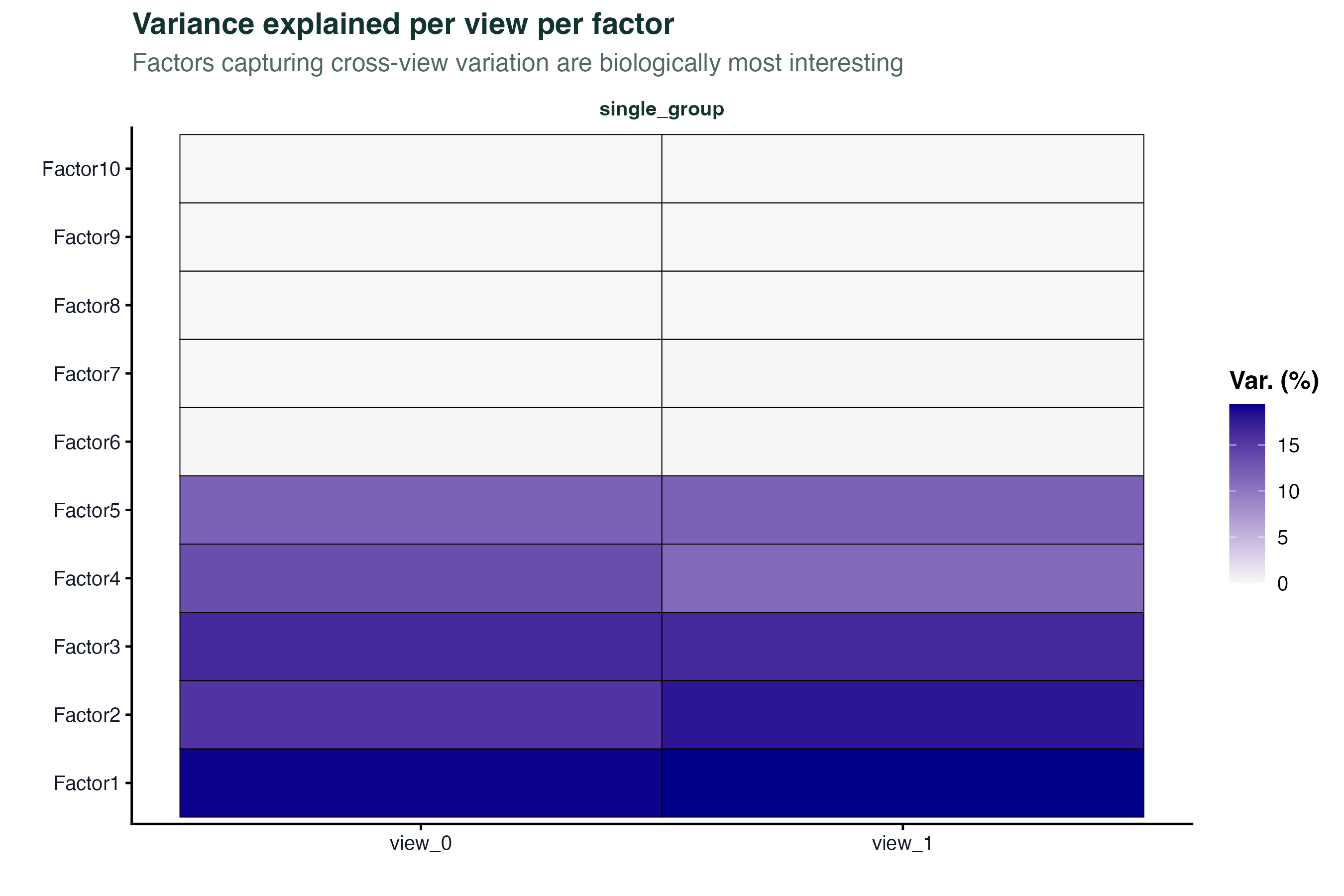
# 热图:行是因子,列是组学层
plot_variance_explained(model, x = "view", y = "factor")
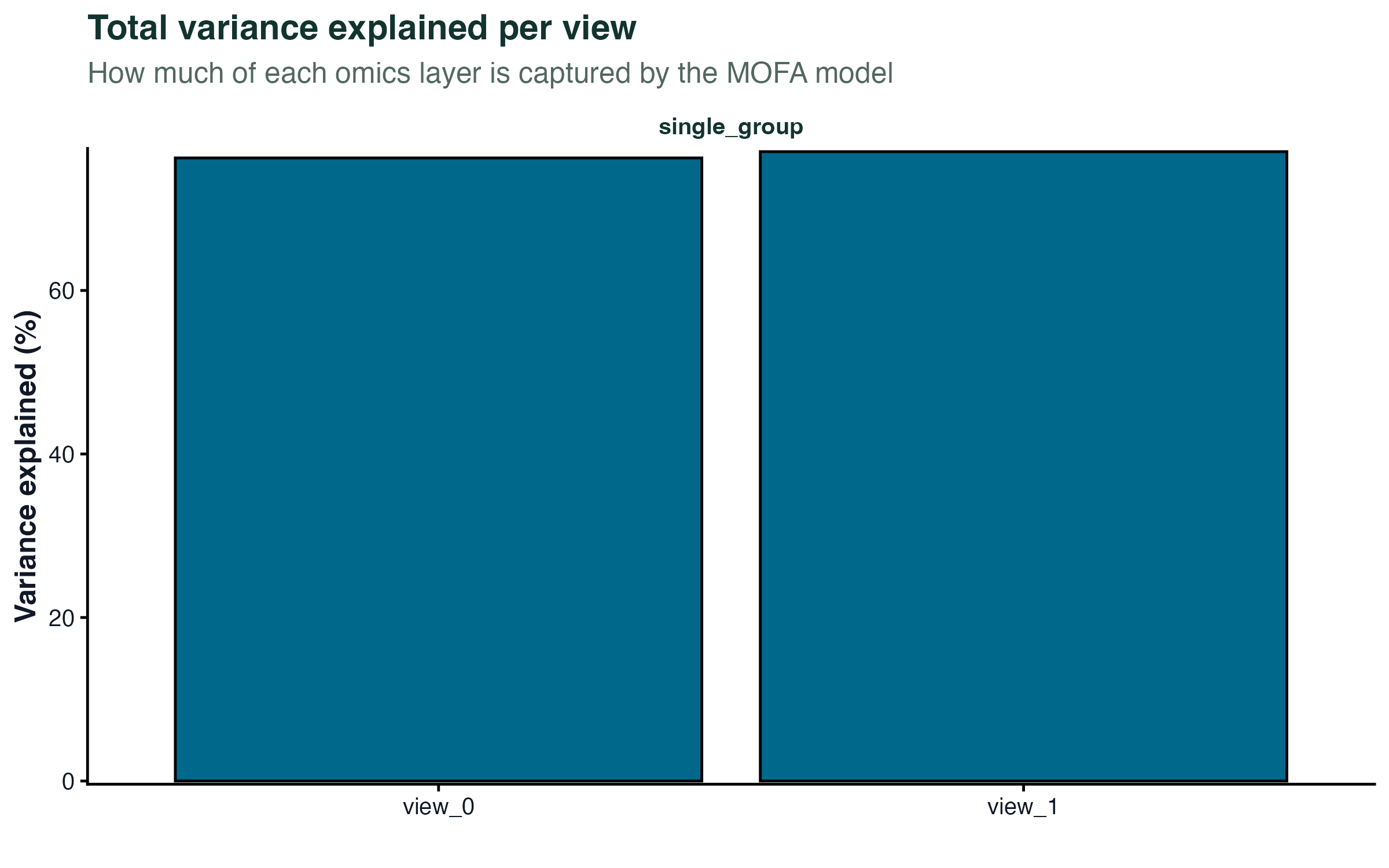
# 每个组学层被所有因子解释的总方差
plot_variance_explained(model, plot_total = TRUE)[[2]]
如果某个因子只在一层有高方差贡献,它反映的是该层特异的变异;如果跨多层都有贡献,它捕捉的是跨组学的共同信号,通常更有生物学意义。
因子权重
权重(weights)告诉你每个因子由哪些特征驱动。权重绝对值大的特征是该因子的核心成员。
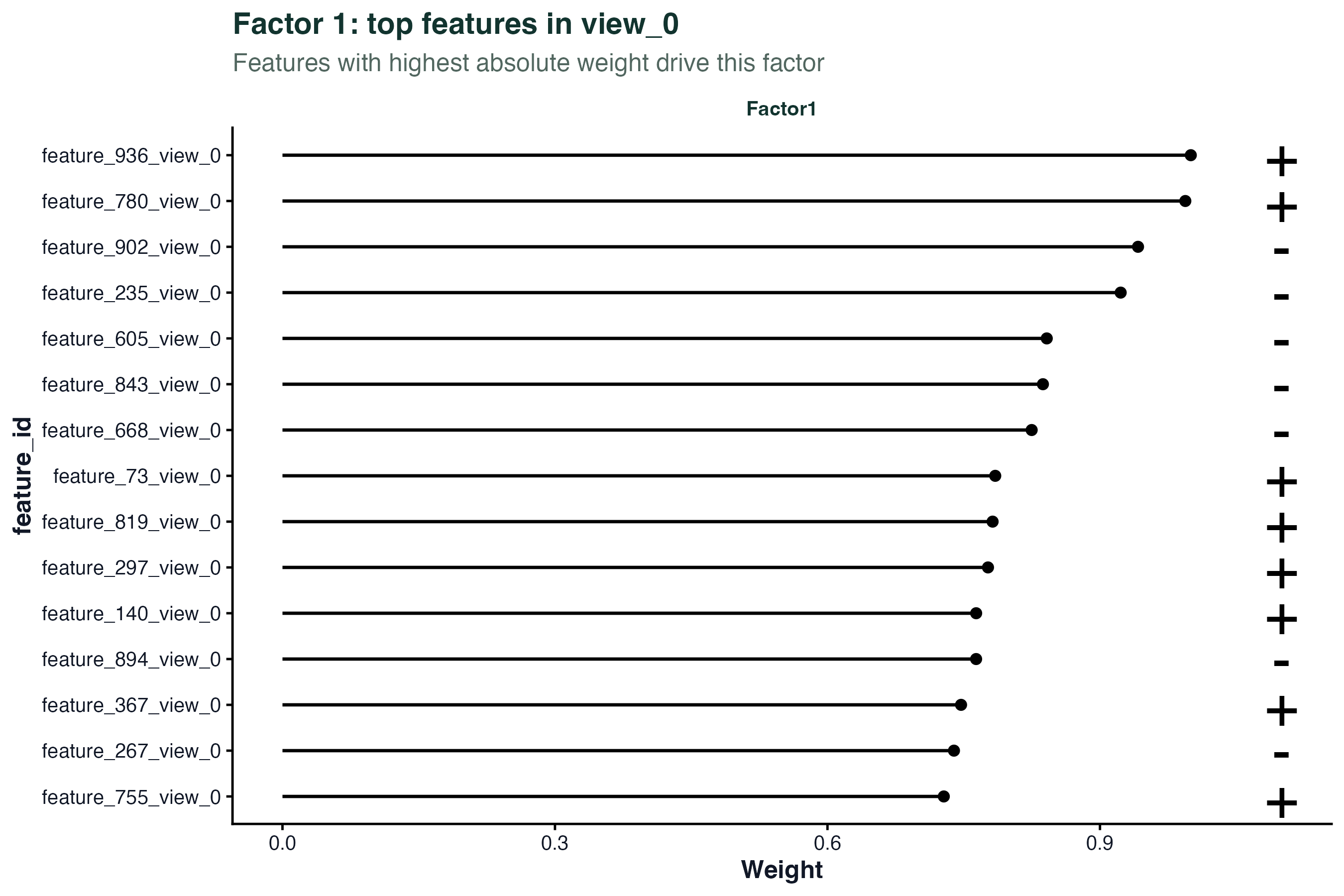
# Factor 1 在 mRNA 层的 top 权重特征
plot_top_weights(model, view = "mRNA", factor = 1, nfeatures = 15)
这些 top 特征可以送去做富集分析(GSEA、GO),看它们是否集中在某个通路。
因子得分与样本分布
因子得分(factor values)是每个样本在各因子上的坐标,类似 PCA 的 score。
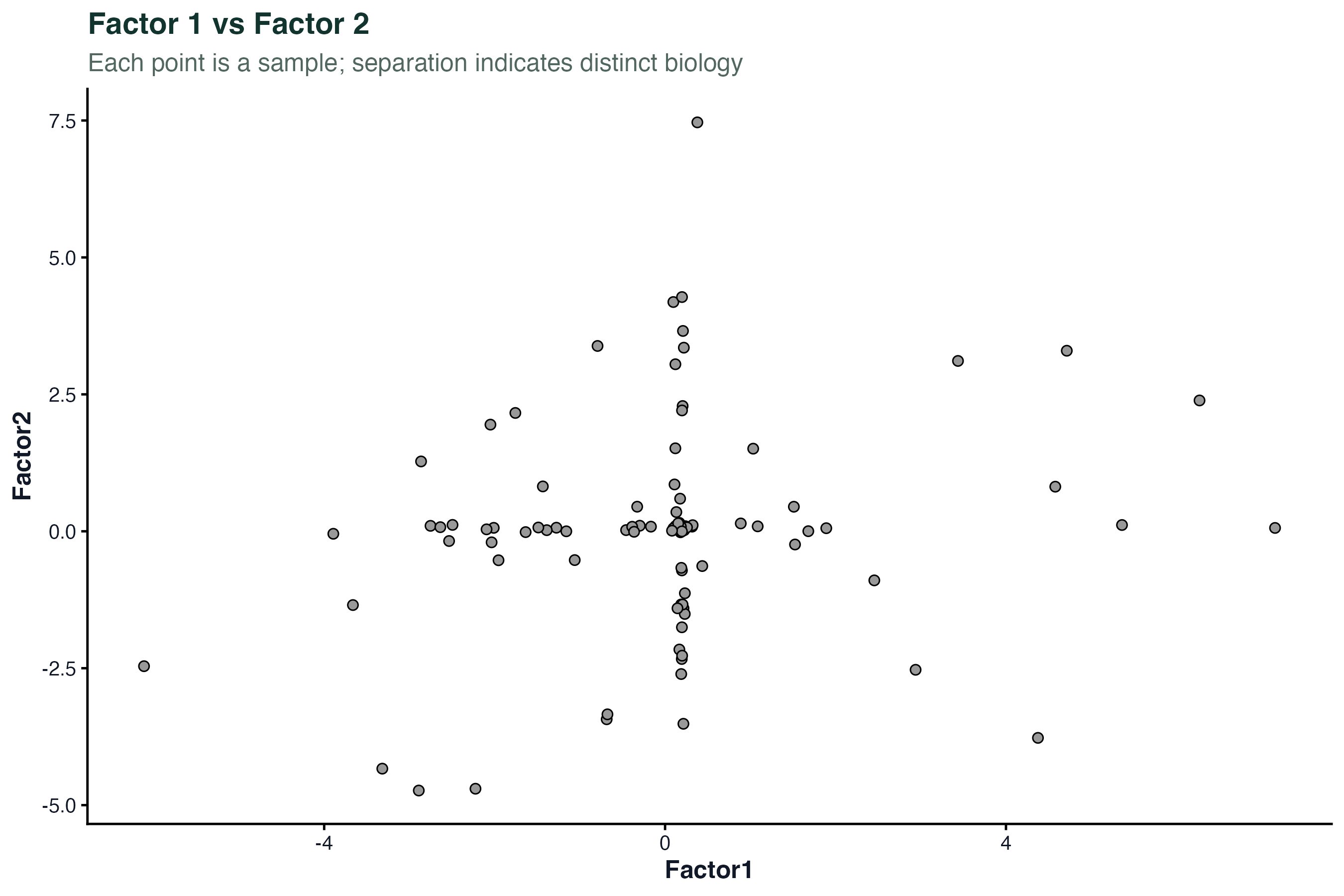
# 用 Factor 1 和 Factor 2 画散点图,按亚型着色
plot_factor(model, factors = c(1, 2), color_by = "IGHV")

# 因子之间的相关性(理想情况下应接近 0)
plot_factor_cor(model)
数据总览
# 展示各层数据的缺失模式和样本覆盖
plot_data_overview(model)
CLL 数据集中 Mutations 层缺失较多,但 MOFA2 能正常处理。
因子的生物学解读
拿到因子权重后,下一步是理解它的生物学含义:
library(msigdbr)
library(fgsea)
# 获取 Factor 1 在 mRNA 层的权重
weights <- get_weights(model, views = "mRNA", factors = 1, as.data.frame = TRUE)
gene_ranks <- setNames(weights$value, weights$feature)
gene_ranks <- sort(gene_ranks, decreasing = TRUE)
# 用 Hallmark 基因集做 GSEA
hallmark <- msigdbr(species = "Homo sapiens", category = "H")
pathways <- split(hallmark$gene_symbol, hallmark$gs_name)
fgsea_res <- fgsea(pathways, gene_ranks, minSize = 15, maxSize = 500)
head(fgsea_res[order(pval), ], 10)
常见坑
坑 1:因子数设太多导致过拟合
把 num_factors 设成 30,结果后面 20 个因子解释的方差不到 1%,既浪费计算又增加解读难度。MOFA2 会自动 drop inactive factors,但初始设太多会让训练不收敛。建议从 15 开始,看方差解释图再决定保留多少。
坑 2:因子解读时只看权重不做富集
Factor 1 权重最高的 10 个基因不一定能代表通路含义。应该把整条权重列表当作 ranked list 送进 GSEA,而不是只取 top 10 做 ORA。后者容易遗漏"多个基因效应小但方向一致"的通路信号。
坑 3:把单层特异因子当作整合发现
如果某个因子只在 Methylation 层有高方差贡献,它反映的是甲基化特异的变异,不是"跨组学整合的成果"。论文里应该重点讨论跨多层都有贡献的因子,单层特异因子可以放到补充材料。
坑 4:没有检查因子的可重复性
换个随机种子重跑模型,如果因子的权重排序完全变了,说明结果不稳定。建议跑 3-5 次不同 seed,检查因子间的相关性(plot_factor_cor)。如果核心因子在多次运行中一致出现,结论才可信。
坑 5:Python 环境缺失导致训练失败
MOFA2 的训练后端是 Python 的 mofapy2。如果 R 找不到正确的 Python 环境(conda/venv),run_mofa 会报错。建议用 reticulate::use_condaenv("mofa") 显式指定,并在 run_mofa 前验证 reticulate::py_module_available("mofapy2")。
下一步
接着深入:
- 06 SNF 相似性网络与亚型识别 — 另一种整合思路:网络融合
- 08 整合结果的生物学解读与报告 — 把因子翻译成论文语言
横向延伸:
- 04 WGCNA 与共表达模块 — WGCNA 模块的 eigengene 可以作为 MOFA 的输入特征
- 蛋白质组 08 与转录组联合分析 — 两层数据的 MOFA 整合实例